Making Rural Communities Visible in Artificial Intelligence Through Rural Proofing in Kansas and Beyond
A road can show connection, but not access. Rural communities might appear in data and public systems, yet still remain invisible when AI systems do not reflect distance, transportation barriers, service gaps, workforce constraints, smaller data sets, and local strengths. Rural proofing gives Kansas and other rural states a practical way to make these realities visible in the AI-driven decisions already shaping health and social services.
Artificial intelligence (AI) is increasingly shaping decisions across public health systems, including how needs are identified, how resources are distributed, and how services are delivered. As a result, AI will play an important role in the future of healthy rural communities. When designed and governed carefully, AI can improve access, resource planning, coordination, and service delivery. When rural contexts are overlooked, AI systems can reproduce uneven outcomes and risk deepening existing disparities. In rural areas, where health systems often operate with fewer providers, thinner infrastructure, and less margin for error (meaning fewer backup resources when something goes wrong), these risks can be especially significant.
This memo examines rural invisibility in AI-related health systems, defined as the underrepresentation of rural communities in data, system design, validation, and governance. It explains why these gaps matter and why AI should be developed, tested, and governed with rural communities in mind. The term “rural” can be defined in a variety of ways, but this memo leans on the shared understanding of rural places as those with fewer people, less population density, and greater distance to services. While each rural community has a different history, strengths, resources and challenges, this memo – and the concept of “rural-proofing”, explained within – recognizes there are many shared challenges commonly faced by rural communities.
At both the national and state levels, there is an opportunity for more intentional action to recognize rural invisibility in AI systems as a policy issue. States can position themselves as proactive leaders in rural AI governance by aligning with federal frameworks while developing practical, state-level approaches. Kansas can become a leader in developing and implementing practical rural-proofing approaches that can serve as a model for other rural states. To do so, the state should take five connected steps: 1) make rural context a required part of any Kansas AI task force; 2) require rural proofing before agencies adopt or expand high impact AI tools; 3) institutionalize rural listening through trusted local partners; 4) document the Kansas model as a public blueprint other states can adapt; and 5) build a statewide rural AI literacy framework for residents,students, frontline workers, and public agencies.
Challenges and Opportunities
Rural communities have strong social connectedness, local knowledge, community leadership, and deep relationships that support resilience and innovation. Yet, they often face lower population density, greater geographic dispersion, and more limited access to services and infrastructure. In these settings, AI decisions in one domain can quickly affect others, making locally grounded context and community-level oversight especially important. As AI adoption grows, its effects on rural communities reach well beyond any single tool or system. What’s at stake is broader: how rural needs are represented in data, who has a voice in how AI decisions are made and governed, and how the benefits and burdens of AI systems and infrastructure are distributed across communities. These dynamics raise important questions about whether AI systems adequately account for rural conditions, populations, and lived experiences.
Rural Invisibility in AI Systems
Rural invisibility in AI systems occurs when rural communities are underrepresented in the data, assumptions, design, validation, and governance that shape how systems are built and used. That can make rural needs harder to see and rural harms harder to detect. In practice, it means that AI systems may be built on assumptions that do not reflect rural realities, leaving rural communities overlooked in decisions about resources, services, and policy.
The body of evidence, including the 2025 scoping review, illustrates how this invisibility carries into practice. It highlights that rural AI research is underdeveloped and that models underperform in rural settings, and the consequences of those failures are rarely studied where they are felt most. As the 2025 National Rural Health Association policy brief notes the challenge is not simply whether rural systems use AI, but whether technologies reflect the realities of fragmented records, thin staffing, and delayed care pathways. When those realities remain invisible in design and implementation, the consequences can include missed, delayed, or incorrect diagnosis, misallocation of resources, and greater strain on rural providers.
Gaps in AI Governance Frameworks
It is important to assess how well current governance approaches perform across different contexts. Current AI governance frameworks, including the National Institute of Standards and Technology Artificial Intelligence Risk Management Framework and Organization for Economic Co-operation and Development (OECD), provide a strong foundation by emphasizing fairness, transparency, accountability, and risk mitigation, but they provide limited guidance on how to operationalize these principles in rural environments. These frameworks often do not fully account for differences across settings. For example, communities and organizations vary in data availability, institutional capacity, and service infrastructure. They also differ in their ability to evaluate and govern AI tools, especially when staffing, technical expertise, and resources are uneven. Most frameworks do not require testing across small or geographically distinct populations, which can make it harder to see how AI performs in rural areas and allow disparities to go unnoticed.
In addition, current frameworks do not specify how local knowledge, professional judgment, or community perspectives, particularly those from rural communities, should be incorporated into AI oversight and decision-making, which can both algorithmic invisibility and broader forms of rural invisibility in AI. While they emphasize stakeholder engagement, they leave implementation largely undefined, which can limit the ability to identify context-specific risk. These gaps also matter because AI already shapes public benefits, legal navigation, housing, and service coordination. When trained on data shaped by past inequities, AI can deepen disparities rather than reduce them. This is why AI governance must move beyond general principles and explicitly incorporate rural proofing, accountability, and meaningful community involvement.
Federal policy remains an important lever because it can help push state policy forward by signaling priorities, shaping governance expectations, and giving states a stronger foundation for action.Current federal guidance provides a foundation for responsible AI use but offers more limited practical direction for rural settings, where sparse data, limited staffing, and fragmented service systems can affect how AI works in practice. Even though the recommendations in this memo focus primarily on actions at the state level, federal guidance on addressing rural invisibility in AI across health, education, and social systems can help create the conditions for states to act more effectively and equitably on behalf of rural communities.
The White House Office of Science and Technology Policy (OSTP) or the Domestic Policy Council (DPC) is well positioned to lead coordination across federal agencies, ensuring that rural AI implementation challenges are recognized in efforts affecting health, education, and social systems. Building on that coordination, the Office of Management and Budget (OMB) is well positioned to reinforce this work through its existing governance and procurement role to clarify how existing expectations for artificial intelligence procurement, validation, monitoring, oversight, and accountability apply in rural-serving settings. The Department of Health and Human Services (HHS), the Department of Agriculture (USDA), and the Department of Education (ED) should then help translate that guidance into practice for artificial intelligence systems and programs that directly affect rural communities. The National Institute of Standards and Technology (NIST) should provide supplemental examples showing how artificial intelligence risks can present differently in rural settings. This would strengthen implementation under existing frameworks without requiring the development of a separate framework.
Federal agencies should use existing programs to strengthen rural data infrastructure, technical assistance, and workforce readiness, and governance capacity needed for responsible AI implementation in rural communities. HHS, USDA, and ED can support rural-serving institutions directly, while NIST and other federal partners can provide tools, guidance and practical examples to help organizations implement AI responsibly and effectively.
The Need for Rural Proofing
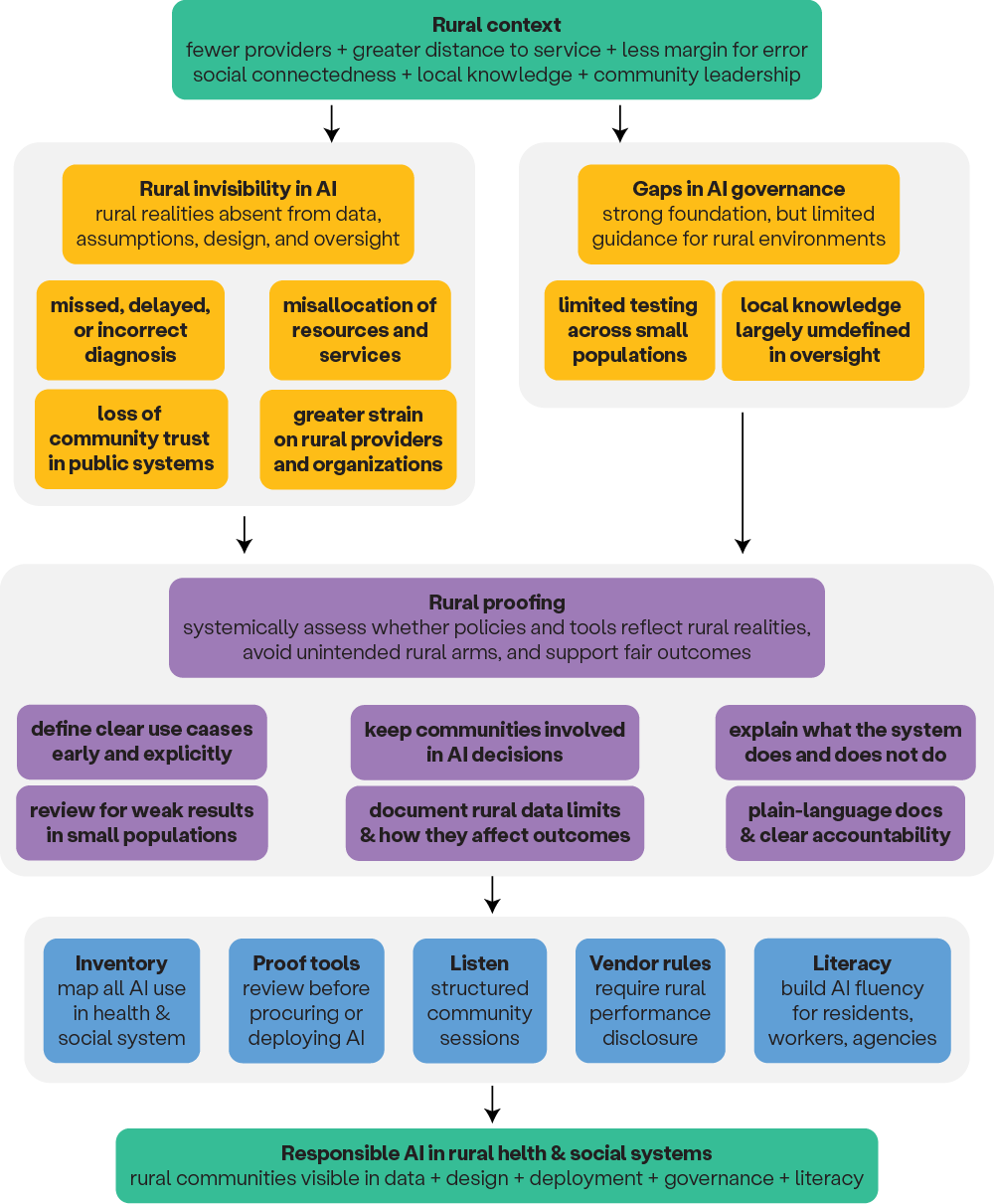
Rural proofing is the process of systematically checking whether policies, tools, and investments reflect rural realities, avoid unintended rural harms, and support fair outcomes for rural communities. In practice, it means asking early and explicitly how a policy or AI system will function in places with lower population density, greater distance from services, thinner infrastructure, smaller administrative capacity, and different patterns of need and service use.
When applied to AI, rural proofing makes rural conditions visible across system design, data, deployment, and oversight. This includes defining clear use cases, keeping communities involved in decisions about AI, explaining what the system does and does not do, and regularly reviewing whether it creates unequal results. It also means regularly reviewing system performance, checking for weak results in small or low-volume populations, documenting when rural data is limited, and being transparent about how those limitations affect outcomes. Rather than treating rural impact as an afterthought, rural-proofing makes rural context and rural strengths a core part of design, implementation, oversight, and evaluation. Within governance processes, it also helps ensure that policies and decisions are informed by rural needs, contexts, and strengths rather than assumptions developed elsewhere.
Because many rural systems operate with limited staff, tight budgets, and shared regional responsibilities, AI governance requirements must be practical. Federal and state agencies should give rural-serving organizations the time, funding, and support needed to review systems, raise concerns, and participate in oversight. They should also provide plain-language documentation so local leaders, frontline staff, and community members can understand how decisions are being made. Finally, rural proofing requires clear accountability. When AI systems cause harm or fail to work fairly in rural communities, agencies and vendors should have a clear process to identify the problem, respond to it, and fix it (see Figure 1).
Plan of Action
Addressing rural invisibility in AI algorithms and systems across health and social sectors requires coordinated national attention and action, including the integration of rural proofing into national AI governance efforts. Because national frameworks often serve as guidance for states, progress at the national level is needed to provide the standards, expectations, and resources that support states in adapting AI governance to their specific contexts.In the meantime, states can begin building their own pathways by aligning with existing frameworks, piloting approaches in priority areas, and strengthening internal capacity.
Kansas as a Blueprint
As one of the nation’s rural states, Kansas has a strong interest in ensuring that AI systems work effectively for rural communities. As AI becomes increasingly integrated into sectors that are important to rural Kansan, including health care, education, transportation, agriculture, emergency response, public benefits, and other public services, rural-proofing can help ensure that AI tools are responsive to rural contexts.
For Kansas, this could include leveraging existing rural health infrastructure, engaging local stakeholders, and testing practical approaches that can be scaled as clearer national direction emerges. The Center for Medicare and Medicaid Services (CMS)’s Rural Health Transformation Program offers one practical pathway for aligning rural technology investment and technical assistance in Kansas with rural AI proofing principles. The Kansas Legislative Artificial Intelligence Task Force should explicitly include rural context as a defined part of its charge, membership, and workplan. The Kansas Office of Information Technology Services (OITS), the Information Technology Executive Council (ITEC), the Kansas Department of Health and Environment (KDHE), the Kansas Department for Aging and Disability Services (KDADS), and the Kansas Department for Children and Families (DCF) should work collectively to translate broad AI governance principles into practical oversight and implementation for rural health and social systems.
Furthermore, implementation of these recommendations can be staged based on current capacity, allowing agencies to begin with foundational actions and progressively build toward a more coordinated, statewide approach over time (see Figure 2).

Recommendation 1. Ensure The Kansas Legislative Artificial Intelligence Task Force and Any Future State-Level Task Forces Explicitly Include a Focus on Rural Context and Health
The Kansas Legislative Artificial Intelligence Task Force, given its role in shaping AI policy and direction, should explicitly include rural context as a defined part of its charge, membership, and workplan. The current task force already includes legislators, executive branch leadership, universities, health systems, agriculture, and private sector technology members. The taskforce’s scope could include reviewing AI use in rural contexts, incorporating rural and frontline voices into decisions around AI procurement and deployment, and issuing guidance on procurement, oversight, and accountability in rural health and social systems.
In practice, Kansas can build on the existing role of OITS by extending its coordination function to include AI-specific responsibilities, such as setting standards for evaluation, interoperability, and responsible use across agencies. ITEC can provide statewide governance direction by aligning AI efforts with broader IT strategy and policy. Service agencies, including KDHE, KDADS, and DCF would implement these efforts within health and social systems. This structure gives Kansas a practical model that other states can adapt by pairing a statewide IT authority with the agencies that directly manage public benefits, care, and social services.
- Define Scope. State-level Kansas AI task forces, working groups, and advisory bodies should explicitly include AI use and governance in rural contexts as a core scope of work, embedding rural considerations directly into the charge, membership, and workplan rather than treating them as secondary or optional.
- Coordinate Governance. Cross-agency coordination should occur through OTIS with statewide governance direction set through ITEC, while agencies including KDHE, KDADS, and DCF should identify and document where AI affects health and social service access.
- Establish Structure. Kansas should establish a clear governance structure that translates national AI principles into practical oversight, procurement, and implementation decisions tailored to rural conditions.
Recommendation 2. Require Rural Proofing for AI Used in Kansas Health and Social Service Programs
AI-enabled tools are expanding across eligibility decisions, care coordination, analytics, and service delivery. Because of this, Kansas should strengthen AI governance within the agencies that directly shape health and social outcomes. In practice, this work should begin with KDHE, KDADS, and DCF with cross-agency coordination support from OITS. Rather than relying only on broad fairness principles, these agencies should use a practical rural-proofing process to assess whether AI tools work reliably in rural settings with different staffing levels, service access, broadband conditions, data volume, and administrative capacity. Taking these steps now would help Kansas clarify oversight responsibilities, procurement standards, and rural risk before AI becomes more deeply embedded in public systems.
- Inventory. KDHE, KDADS, and DCF should identify and document current and planned uses of automated decision tools and AI systems used in decision-making that affect eligibility, benefits, care access, case management, service coordination, navigation, and enforcement. Agencies should require vendors to inventory AI used in care management, prior authorization, utilization management, member outreach, and provider network decisions. Kansas can then expand this inventory approach to other agencies that shape health, including housing, transportation, workforce, education, justice, and environmental systems.
- Rural AI Proofing. Agencies should apply rural-proofing review before they procure, renew, expand, modify, or deploy high-impact AI systems. This review should assess whether the tool performs reliably in rural settings, whether rural data are sufficient for validation, whether lower service use is being misread as lower need, and whether the system creates added burdens in places with limited staff, broadband, transportation, or service infrastructure.
Governance and Coordination. Kansas should establish a centralized cross-agency approach to AI governance to ensure consistency and avoid fragmented implementation across agencies. A coordinated structure—led jointly by OITS and state procurement—should define statewide oversight, reporting expectations, and minimum standards for the use of AI and automated decision tools. - Strengthen and Formalize AI Governance. Within this structure, agencies should strengthen and formalize AI governance by assigning clear oversight responsibility, documenting risk management and vendor review practices, incorporating transparency and explainability requirements, and building internal and community-level AI literacy. Agencies should also implement consistent oversight and reporting expectations for vendor AI use, including requirements for rural proofing, audit, and review mechanisms. OITS, in coordination with state procurement, should provide cross-agency technical guidance, ensure consistency in procurement standards, and align AI use with state IT and data governance policies. Individual agencies should retain program-level oversight within their statutory authority while operating within this coordinated governance framework.
- Vendor Requirements. Require vendors to explain how their AI tools perform in rural settings, disclose known data and performance limits, identify human review points, and provide plain-language documentation on system purpose, intended use, and potential failure points.
Recommendation 3. Institutionalize Rural Listening through Trusted Intermediaries
Meaningful engagement with rural communities is especially important in this context because AI systems are often designed and evaluated far from the places where their effects will be felt. However, engagement alone is insufficient. This recommendation draws a deliberate distinction between consultation, where agencies ask communities what they think, and co-governance, where rural communities hold real influence over AI decisions that affect them. Kansas should aim for co-governance, not just input collection In rural areas, where access to care, public services, transportation, broadband, and legal support may already be limited, even small design flaws or inaccurate assumptions can have outsized consequences. Regular listening with rural residents and trusted local partners can help surface needs, barriers, and unintended harms that may otherwise remain invisible in statewide decision-making.
- Support. OTIS or the Governor’s Office should coordinate recurring AI listening sessions, with participation from KDHE, KDADS and DCF, and with research and facilitation support provided by Kansas public universities.
- Leverage Partners. OITS and KDHE, KDADS, DCF should engage Kansas public universities to support session design, facilitation, documentation, synthesis of findings, and evaluation to ensure structured feedback and continuity across sessions.
- Conduct Sessions. KDHE, KDADS, DCF in partnership with county and local public health agencies, behavioral health providers, university extension networks, libraries, legal aid and court self-help programs, and community-based organizations, should conduct sessions using a shared calendar and unified feedback process.
- Apply Findings. KDHE, KDADS, DCF should use listening sessions as a rural-proofing mechanism to assess how AI-enabled tools in health, benefits, and social service programs affect access to care, legal protections, and social determinants of health in rural settings.
- Integrate Oversight. OITS and KDHE, KDADS, DCF should integrate findings into state oversight by documenting recurring rural issues, flagging systems for review, and using insights to strengthen procurement, monitoring, and accountability for AI systems used in Kansas programs.
Recommendation 4. Establish a Kansas ‘Rural AI Health Governance Blueprint’ for Other Rural States to Replicate
Clear leadership at the state level matters because rural proofing is unlikely to be applied consistently if agencies and vendors are left to interpret it on their own. A statewide approach creates shared expectations, strengthens accountability, and makes clear that rural context should be built into procurement, oversight, and evaluation from the beginning. This approach is also replicable because it relies on documented processes, practical tools, review steps, and implementation lessons that other rural states can adapt to fit their own governance structures, service systems, and community conditions. The framework should also incorporate AI infrastructure impacts, including data center siting, to ensure rural-proofing standards address the distribution of resource, environmental, and land use burdens associated with AI development.
- Document Framework. OITS, in coordination with the Information, ITEC and participating agencies such as KDHE, KDADS, and DCF, should document Kansas’s rural AI governance framework in a public implementation guide, including rural-proofing standards, shared workflows, listening session models, and transparency practices.
- Assess and Evaluate. KDHE, KDADS, DCF should partner with Kansas public universities to assess outcomes, identify lessons learned, and produce evidence-based recommendations to strengthen rural AI governance over time.
- Share and Scale. OITS and participating agencies should share implementation tools, templates, rural-proofing checklists, and model policies through interstate networks such as the National Governors Association, the National Conference of State Legislatures, and state rural health associations so other states can adapt the Kansas model.
- Pilot Collaboration. OITS, with support from Kansas public universities and partner agencies, should pilot cross-state collaboration with at least two predominantly rural states to test whether the Kansas workflow can transfer across different governance structures, agency arrangements, and service systems.
- Demonstrate Practice. OITS, state service agencies, and university partners should position Kansas as a practical demonstration state by showing how rural proofing can translate broad AI governance expectations into workable state practice that reflects rural conditions, administrative limits, and community realities.
Recommendation 5. Establish a Standardized and Contextualized Kansas Rural AI Health Literacy Framework
Kansas should complement the upstream AI governance framework with a statewide Rural Health AI Literacy Framework to ensure residents, students, and frontline workers can engage AI systems critically. Unlike general AI literacy, which often focuses on basic awareness of AI tools and digital skills, rural health AI literacy should prepare residents, students, frontline workers, and public institutions to understand how AI can shape health access, eligibility, referrals, triage, service coordination, and related decisions in rural communities. Governance structures alone are insufficient if communities lack shared standards for understanding how AI affects eligibility, health access, agriculture, transportation, and legal services in rural settings. The Kansas State Department of Education (KSDE), in coordination with the Kansas Board of Regents (KBOR) and the Kansas Office of Information Technology Services (OITS), should lead development of tiered, age-appropriate AI literacy competencies spanning K–12, postsecondary education, and public-sector roles.
To operationalize this framework, Kansas should:
- Integrate Curriculum. KSDE should integrate AI literacy into digital literacy, civics, agricultural education, and career and technical education standards, with support from Regional Education Service Centers for teacher training.
- Embed in Higher Education. KBOR should embed AI literacy modules into general education requirements and first-year seminars across public universities and community colleges, aligning with Higher Learning Commission expectations.
- Establish Rural Hubs. Kansas State University and Cooperative Extension should serve as rural AI literacy hubs delivering applied programming for health, agriculture, and local government sectors.
- Expand Community Delivery. State agencies and education partners should collaborate with public libraries, Tribal education departments, workforce development boards, and community-based organizations to deliver multilingual AI literacy programming statewide.
- Train Public Workforce. State agencies should implement baseline AI literacy training for employees in health, eligibility, and human service roles.
Conclusion
As AI becomes more embedded in public systems affecting health and social outcomes, it is important to account for rural context, particularly in Kansas, where many communities operate under conditions that differ from those assumed in typical AI development and deployment environments. These conditions include greater data sparsity, lower service density, and constrained institutional capacity for oversight. The proposed recommendations aim to operationalize responsible AI principles through coordinated cross-agency governance, integration of rural proofing into existing structures, and stronger community engagement in AI decision-making. By acting now, Kansas can build a more accountable model for rural AI governance and offer other rural states a practical path forward.
Rural health refers to the health outcomes, service access, and community conditions that shape well-being in rural communities. It includes access to healthcare, behavioral health, substance use treatment, prevention, workforce capacity, transportation, and the social determinants of health that affect whether rural residents can receive timely and appropriate care.
Common federal rural definitions include those developed by the U.S. Census Bureau, the Office of Management and Budget, and the U.S. Department of Agriculture Economic Research Service. The ideas, challenges and recommendations presented here within, but are not limited by, common rural definitions used across public health and health care. While rurality exists on a spectrum, definitions often use some combination of population thresholds, population density, housing density, and proximity to dense urban areas to define levels of rurality and urbanicity.
They should establish AI governance structures and policies, inventory current and planned AI use, assess whether tools are necessary and can function effectively in rural settings, document rural data limitations and oversight responsibilities, require vendor disclosure, and provide plain-language information about how systems work and how human review and oversight are incorporated into decision-making.
AI vendors should explain how their systems perform in rural settings, disclose known data and performance limitations, identify human review points, and provide plain-language documentation on system purpose, intended use, and conditions under which performance may vary.
Listening sessions help state agencies hear directly from rural residents, frontline workers, and local organizations about how AI affects access to care, benefits, legal navigation, and other services in practice. The memo recommends using those findings to improve procurement, monitoring, and accountability.
Public Participation IS the Ingenuity We Need
Building Blocks To Make Public Participation Solutions Work
Participation is not a distraction from governing — it is how government governs well. When treated as compliance, it comes too late and excludes those most affected, weakening legitimacy. Designed as a strategic asset, it builds trust, eases implementation, and supports more durable decisions.
Implications for democratic governance
- Participation is how decisions are made. Engagement should focus on clearly defined choices, constraints, and tradeoffs so decisions can move forward.
- Who participates, and when, shapes what government hears. Participation must reflect the full scope of impacts, not just the most visible or organized voices.
- Legitimacy depends on follow-through. Agencies should explain how input was considered so people can make sense of the outcome, even when they disagree.
Capacity needs
- Learn and adapt throughout the process. Track who is participating, what input is generated, and how it is used — then adjust the approach as needed.
- Engage early enough to matter — and continuously where possible. Bring people in while options are still open, and stay engaged so feedback informs both decisions and implementation.
- Structure input for decisions. Ask targeted questions and use formats that help compare impacts, priorities, and implementation considerations.
- Train for real-world engagement. Equip staff to facilitate conversations, synthesize input into key themes, and navigate disagreement.
- Make participation feasible. Offer enough lead time, plain-language materials, and multiple ways to engage (e.g., virtual, in-person, asynchronous) that account for different access needs.
Government, at its best, is democracy’s promise made real: the mechanism through which a society turns values into action and public voice into policy. But that mechanism has corroded. Rebuilding it starts with something simple — treating the public not as a problem to manage, but as a source of ingenuity government cannot function without.
Public participation, as the federal government executes it today, rarely builds trust: the public hearing held after decisions are already made, the comment period that produces thousands of responses with no visible impact, the listening session where officials take notes but never engage. The other version, where a highly organized few monopolizes public ears and distorts public response for their niche interests, is equally demoralizing. Critics are right to call out this failure. Across the political spectrum, there is a shared diagnosis: the current system of public engagement too often functions as a series of veto points, rewarding obstruction over problem-solving and delay over delivery.
That erosion matters enormously right now. Americans have grown deeply skeptical that government can solve hard problems, and climate change, with its complexity and its demands on every sector of the economy, may be the hardest challenge it has ever faced. As the authors ask in the opening argument of the Center for Regulatory Ingenuity, if government can’t effectively address challenges it deems an “existential threat,” what good is it, and can democracy overcome this downward spiral of mutually reinforcing cynicism?
We believe it can. Today, we are living through a mid-transition moment in climate policy, in which the technologies we need exist, the economics are increasingly favorable, and many obstacles are governmental: slow processes, fragile coalitions, and policies that get built and then litigated into irrelevance. In that context, the instinct to streamline is understandable. But a government that privileges artificially weighted listening, or avoids listening because it didn’t plan well, doesn’t move faster. It moves blindly or with bias. It builds the wrong things, in the wrong places, for the wrong reasons, and then wonders why nothing sticks. The argument here is not for more or less participation but for better participation, treated as a strategic asset rather than a box to check, and designed with the same rigor as any other policy instrument. Done well, public engagement demonstrates that government can listen, adapt, and earn trust. At a moment when democratic institutions are fragile, that demonstration is not incidental to the work of governing, it’s central to it.
Climate change is not just a technical problem, it’s a governance problem, and increasingly, a democratic one. It reaches into every community, every economy, and every aspect of daily life, from how people power their homes to how they move through their cities to whether their communities remain livable at all. There is no technocratic solution that can bypass the public. The choices climate demands (where to build, what to prioritize, who bears costs, and who benefits) are fundamentally democratic choices. If democratic government cannot make those choices in ways that are effective and legitimate, it will not just fail on climate. It will fail at its most essential purpose: helping people shape the conditions of their shared future.
Definitions and the Role in Governance
Before making the case for better participation, it helps to be precise about what we mean in a space where a range of practices often get conflated.
In January 2025, the White House Office of Management and Budget issued a memorandum that the authors of this paper helped develop, laying out a federal framework for broadening public participation and community engagement. The memo itself was built through the practices it recommends: a public request for information drew input from hundreds of participants and nearly 300 written comments, documented in a public summary that informed the final guidance. It offers definitions worth building on. Public participation is any process that engages the public in government decision-making — helping shape policies, regulations, or research, or soliciting new ideas and innovations. It is inherently transactional: it seeks to inform and obtain input from those interested in or affected by agency action. Community engagement, by contrast, is primarily relational — the consistent building of relationships with communities over time, informed by the history those communities have with an agency, and transparent about the real opportunities and real limitations of that relationship.
Public participation without community engagement can produce processes that feel hollow — technically open, but not genuinely accessible to the people most affected. Community engagement without public participation can build trust and goodwill that never translates into actual influence over decisions. Together, they reinforce each other: relationships make meaningful input possible, and meaningful input, when reflected in decisions, strengthens those relationships over time. But they are not interchangeable, and not always needed in equal measure. One of the memo’s most important contributions is helping government actors think more carefully about which tool fits which moment; a public comment period serves a different purpose than a years-long relationship with a frontline community, and conflating them produces both bad process and bad outcomes. Resources like EPA’s Public Involvement Spectrum make this concrete, mapping participation options from basic outreach through information exchange, recommendations, agreements, and stakeholder action, each carrying a different promise to the public and requiring a different level of agency commitment and design.
What the OMB memo does is not add new mandates (federal statutes from the Administrative Procedure Act to the National Environmental Policy Act already require it across a wide range of agency functions). It strengthens and clarifies what good practice looks like within those existing requirements while making that guidance accessible to anyone in government who wants to apply it beyond those requirements.
That the memo exists at all reflects a recognition that the statutory floor was never enough and democratic practice does not end on election day. The case for meaningful participation rests on the idea that government derives its legitimacy from the people it serves, and that legitimacy has to be earned continuously.
Why Engagement Often Fails
If the case for participation is so strong, why does it so often fail to deliver? Not because the theory is wrong! But because the dominant models were built for a different era, and even well-intentioned efforts, when filtered through broken processes, can deepen the cynicism they were meant to address.
This matters because bad engagement actively fuels the cynicism it was supposed to address. Every bad process reinforces the conviction that participation is theater (or participation is illegitimate, or corruption sanitized) and makes it harder to do the real thing next time. These failures matter in any policy domain, but for climate they are something closer to existential — not just for policy outcomes, but for faith in democratic governance itself.
The Design Failures Are Structural, Not Incidental
The most common problems with public engagement are baked into the dominant models. Traditional notice-and-comment processes, for instance, were designed for transparency, judicial review, and weighting technical and legal expertise over lived experience — and they achieve those aims in a narrow procedural sense. But transparency is not the same as accessibility, and access is not the same as influence. As Nicholas Bagley has argued, procedural rules now actively exacerbate the very problems they were designed to solve. Notice-and-comment tends to reward the organized, the well-resourced, or the professionally represented, producing voluminous records that reflect the priorities of those who could afford to engage, not necessarily the communities most affected by the decision. A single well-funded group can submit thousands of pages of technical comments; a frontline community facing the same decision may have no idea the comment period exists, and no capacity to respond to it even if they did.
Formal public hearings share similar pathologies. They are designed to create a record, not a dialogue, and they tend to produce exactly what their format invites. Participants deliver prepared statements into a microphone. Agency officials listen without responding. The atmosphere is frequently adversarial, structured in ways that entrench “us versus them” dynamics rather than creating any genuine opportunity for exchange, learning, or compromise. People leave feeling unheard not because their words weren’t recorded, but because nothing about the process suggested anyone was actually listening.
One-way communication runs across many engagement formats. As the IAP2 Public Participation Toolbox makes clear, even well-designed information-sharing tools — fact sheets, websites, press releases — have significant limitations when they substitute for genuine dialogue rather than supporting it. Listening sessions, informational webinars, and town halls designed primarily to transmit information leave the public as a passive audience (or unheard correspondent) rather than active participants. When people cannot ask questions, push back, or engage in real exchange with the decision-makers who affect their lives, the engagement reinforces rather than reduces the distance between government and community.
Timing compounds all of these design problems. Engagement that happens too late in the decision-making process — after the key choices have been made, after alternatives have narrowed, after political and financial commitments are in place — is unlikely to meaningfully influence outcomes no matter how well it is conducted. Collaboration works best when it begins early, when there is still genuine room to shape the purpose, alternatives, and design of a proposed action. By the time a formal public comment period opens on many major decisions, the substantive work is essentially done. Community input at that stage can affect the margins but rarely the fundamentals and communities know it. When people show up, offer input, and watch nothing change, they stop showing up.
The scale and complexity of engagement materials create their own barrier. Technical documents running hundreds of pages, written in regulatory language accessible only to specialists, are not a neutral feature of the process — they are a filter. Effective strategic communication requires understanding who the audience is, what they already know, and how the issue connects to their lives, none of which is served by dense regulatory documents distributed through official channels. When the baseline requirement for participation is fluency in administrative law and agency-specific jargon, the people best positioned to engage are lawyers and lobbyists, not the residents of a community downstream from a proposed facility. Simplifying materials is not dumbing them down. It is recognizing that the expertise of affected communities is just as relevant as the expertise of credentialed professionals — and that accessing it requires meeting people where they are, not where the agency finds it convenient.
Perhaps most corrosively, some procedural tools originally designed to protect communities have been repurposed as obstruction. When participation requirements become veto points and when the primary function of an engagement process is to create grounds for litigation rather than to genuinely improve decisions, they undermine both the efficiency that critics of government rightly demand and the democratic accountability that participation is supposed to deliver. Small, organized, well-resourced groups can exploit procedural requirements to delay or block decisions that broader communities support, effectively capturing processes intended for the many and wielding them on behalf of the few. This is the participation failure that most directly drives the “just build it” impulse and it is a legitimate grievance. A system that was designed to give communities a voice has, in too many cases, been captured, producing neither democratic legitimacy nor efficient delivery. At the same time, when communities are left out of upstream planning, these veto points often become one of the only avenues available to influence decisions.
The Barriers to Inclusion Run Just as Deep
Even when processes are reasonably well-designed, they often fail to reach the communities that matter most. The gap between who participates and who is affected often follows the contours of existing inequality with uncomfortable precision.
Awareness is the first and most basic barrier. Many people simply don’t know that an opportunity to engage exists. Believe it or not, federal agency websites are not where most Americans spend their time, and the Federal Register is not how most communities learn about decisions that will affect them (even for experienced users). When engagement opportunities are announced through official channels that already skew toward the educated, the connected, and the English-proficient, the resulting participant pool reflects those skews. Compounding this is a lack of clarity about what participation is even for: many people who are aware of an opportunity don’t understand how their input could make a difference, or whether it ever has. That uncertainty is itself a barrier, and it is one that agencies rarely address directly.
The materials and communications that agencies use to invite and support participation often create their own exclusions. Technical documents written for specialists, notices distributed through unfamiliar channels, comment periods with deadlines that give working families no realistic time to respond, engagement formats that assume reliable broadband and digital literacy — none of these are neutral design choices. A sound public engagement plan starts by understanding audience needs and building authentic, reciprocal relationships — the opposite of defaulting to formats that are convenient for the agency. Each design choice narrows the pool of who can meaningfully participate, and the cumulative effect is systematic. When engagement materials are only available in English in communities where many residents speak other languages primarily, the process has already decided who counts. When comment periods close before community organizations have had time to mobilize their members, the timeline has already decided who counts. These are decisions that agencies make, often without fully recognizing them as decisions at all.
Physical access barriers operate similarly. Transportation costs, distance to venues, the inaccessibility of meeting spaces for people with disabilities, the difficulty of attending a weekday hearing while working multiple jobs — these are all practical obstacles that might seem minor in isolation but compound into systematic exclusion for communities that are often most directly exposed to the environmental and infrastructure decisions being made. Virtual engagement has opened some of these doors, but it has closed others: digital access gaps, limited bandwidth in rural and low-income communities, and the particular challenges of meaningful online participation for older adults or those with limited technology experience mean that remote options solve some access problems while creating new ones.
Trust may be the most intractable barrier of all, because it cannot be addressed by logistical improvements alone. Communities that have experienced past harms from government (broken promises, extractive processes, decisions that hurt them and were made without them) carry a rational skepticism about whether this time will be any different. That skepticism is not ignorance or apathy. It is an accurate reading of a track record. Overcoming it requires more than a well-designed meeting or a plain-language summary. It requires demonstrated consistency over time: showing up when there is no immediate decision to be made, acknowledging historical harms directly rather than implicitly, and following through on commitments in ways that are visible and verifiable. This is precisely what Hollie Gilman and Sabeel Rahman mean when they argue in Civic Power that meaningful participation isn’t ultimately about better meetings — it is about redistributing power so that those closest to the problem are genuinely part of the solution.
Privacy concerns add another dimension that is easy to overlook from a position of power or privilege. In communities with historically complex or adversarial relationships with government – such as those experiencing overpolicing, immigrant communities, or other underserved groups – the act of identifying oneself in public participation can seem risky. Showing up on the record or speaking at a public hearing can feel less like civic engagement and more like exposure. These are not irrational fears; they reflect lived experience and how government has used information, access, interests, and related levers against communities. Genuine participation in these contexts will not occur just by invitation. Agencies must actively and continuously address the conditions that make showing up feel unsafe (e.g., by using intermediaries, anonymous or aggregated input, or other protective measures) and acknowledge the prior harm.
One additional factor shapes who participates and whether their participation endures: organization. Even when agencies improve conditions for participation, durable community voice does not emerge automatically; it depends on collective capacity, especially in communities historically marginalized by race or class. Without organizational support, individuals often lack the connections, information, and trust to translate lived experience into effective engagement. Organization helps aggregate that experience, sustain engagement, and convert it into usable input for government. In its absence, participation remains fragmented, reinforcing the advantage of those already organized and resourced.
The Benefits of Effective Engagement as a Strategic Asset
In the early 2000s, a proposed bridge crossing the St. Croix River (a National Scenic Riverway on the Minnesota-Wisconsin border) had been stuck in gridlock for five decades. When serious planning had kicked off in the 1980s and 1990s, the 1931 structure was already fracture-critical. Stakeholder groups and a disparate range of public institutions, representing sharply conflicting interests and mutual distrust, had reliably blocked every attempt to move forward.
What finally broke the logjam wasn’t a better technical study or a more powerful agency directive, a least common denominator solution, or a decision to simply ignore the interests of a set of stakeholders. It was a deliberate shift to structured collaboration rather than the usual method of talking at one another with concerns, with no means of finding points of alignment, resolution, or tradeoffs. Twenty-eight stakeholder groups shaped the bridge’s location and design, with a comprehensive mitigation package addressing the natural, social, and cultural impacts of the new bridge. Moreover,
Relationships and communication among the stakeholders improved remarkably during the problem-solving process. In the words of one stakeholder, “We were able to spend the time necessary to get over our natural inclination to not trust people from the other side. […] We had enough time and enough space to come to a conclusion that everybody could feel comfortable with.”
That outcome wasn’t the result of exhaustion, but of deliberate design and an emphasis on negotiation rather than government serving as an answering machine that never calls back.
Before making that case, it is worth naming what good engagement is not. Smart, well-designed participation is not a mechanism for local communities to veto decisions that serve broader public interests. When a neighborhood is asked whether it wants new apartments, or a community facing a new transmission line is simply asked whether they approve, the answer is predictably no — and designing processes that guarantee that outcome is not good participation! It is a design failure that produces exactly the NIMBY dynamics that rightly frustrate those who want to build. The problem is not that people oppose things. It is that poorly designed processes over-sample the most proximate opposition, structure engagement queries without a sense of purpose or audience, start with technicalities rather than longstanding trust, avoid the potential for early negotiation, or systematically exclude the regional, national, and future stakeholders who have just as much at stake in the outcome. These failures aren’t inevitable.
Good engagement produces better decisions.
Agencies have technical expertise, legal authority, and institutional knowledge, but they routinely lack on-the-ground understanding of how policies will actually land in specific communities, what tradeoffs matter most to the people affected, and what solutions might work that no one in a headquarters office has thought of yet. Meaningful participation fills those gaps.Well-designed engagement generates solutions that are more effective, empowers people from different backgrounds, and builds the local networks that make implementation actually work. It brings in lived experience that data alone cannot capture, surfaces local knowledge that improves policy design, and produces decisions that are more responsive to the full range of affected interests rather than the loudest or most proximate ones.
Research on structured deliberation consistently shows that when people are given good information and genuine opportunity to reason together — rather than just reacting to proposals that feel threatening — they reach more nuanced, durable conclusions than either polarized public opinion or top-down expert judgment alone produces. Deliberation, in this sense, is not just a democratic value. It is a technical tool for overcoming the polarization that makes hard policy decisions feel impossible. It is also, critically, a tool for helping people reason past their immediate self-interest toward a broader understanding of tradeoffs, which is precisely what is missing when participation processes sample only those with the most to lose from a particular change, rather than those with the most at stake in the outcome. Done well, it changes which voices dominate and acknowledges the role of power. That shift is the difference between a participation process that ratifies the preferences of whoever showed up and one that actually informs a decision. This is not an argument for endless input or process without limits. As James Goodwin argues in this collection, the most effective participation is targeted rather than open-ended, focused on the core disputes that actually need resolving, rather than generating voluminous records that obscure more than they illuminate. The goal is engagement that is both more inclusive and more purposeful: asking the right questions of the right people at the right time (which means going well beyond the immediate neighborhood to find the people whose lives will be shaped by a decision).
But asking the right questions requires doing the work before the room fills up. Good engagement doesn’t begin with an open-ended invitation to say whatever comes to mind. It begins with agencies doing enough homework (with experts and communities) to frame the problem clearly: what is actually being decided, what constraints are real, what tradeoffs exist, and where there is genuine room for public input to influence the outcome. That frame is one of mutual respect.
This is where standards matter, as explored later in this essay. The difference between engagement that produces insight and engagement that produces noise is largely a design question.
Good engagement builds the trust that makes government function.
Scholars have considered the consequences of low trust through many lenses, but the legitimacy of democracies relies on trust. Lower trust means less engagement with functions that government performs uniquely or drives, whether disaster response, weather warnings, federal benefits, security functions, public health, or independent data collection and analysis — and that lesser engagement means those functions work less well for everyone else. This has a cascading impact as democratic institutions weaken when government cannot, does not, or is not believed to deliver on expectations of its citizens.
When people experience decision-making as transparent, accessible, and genuinely responsive to their input, trust builds. When they show up and feel unheard, or never show up at all because no one made it possible, trust erodes, most quickly and most consequentially for those communities that can least afford to lose it. This isn’t incidental — trust is the medium through which every other benefit of engagement operates. When it erodes, the downstream consequences reach far beyond any single process, a pattern worth examining directly when we turn to why engagement so often fails.
While study after study shows that less than half of Americans trust the federal government, far fewer (21%) believe it listens to the public — and just 15% believe it is transparent, both according to a 2024 national survey. This underscores a deep perception that government is neither responsive nor accountable to the people it serves. Every engagement process is a small test of whether democracy is meaningful — especially when government is asking people to accept changes as visible and consequential as those required by climate policy.
Good engagement reduces conflict and can prevent litigation.
As Andy Gordon has argued at FAS, listening is a prerequisite for discovery and a requirement for success on any ambitious public goal where stakes are high. The ARPA-I national listening tour demonstrated this concretely: starting with questions rather than answers, and drawing on distributed expertise from every layer of the transportation system, produced an agenda-setting process that no small group behind closed doors could have replicated. The same principle holds for climate and environmental policy. The instinct to streamline participation (to save time, to avoid change, to avoid the NIMBY trap) often backfires in the most concrete terms. Decisions made without adequate input tend to generate opposition downstream, when it is far more costly to address: through litigation, organized resistance, implementation failures, and the kind of sustained community distrust that shadows projects for years. The veto-point problem, in other words, is not a feature of too much participation. It is what participation looks like when it arrives too late, is structured too adversarially, and samples too narrowly. Fix the design (broaden who is heard, start earlier, frame the problem honestly) and participation stops functioning as a veto mechanism and starts functioning as the evidence base that allows government to make hard calls with confidence. An agency that has genuinely sought broad, representative input is in a far stronger position to defend a difficult decision. When that engagement runs through organized, representative groups, it also enables more effective deal-making — where tradeoffs can be negotiated and outcomes reflect a broader, more representative set of community interests.
Collaborative approaches improve the quality of decision-making and increase public trust precisely because they bring the right stakeholders in early (engaging more upstream), before positions have hardened and before the public record has closed. Agreements get built earlier, key voices feel heard rather than steamrolled, impacts are better understood across not only the loudest but those most widely impacted, and mitigation is transparent. And with that, the potential litigation drops, compliance improves, and implementation becomes something communities feel invested in rather than something done to them.
A Framework for Doing It Right
Moving with the public is how government earns trust and makes decisions people can understand, accept, and stand behind.
Participation doesn’t eliminate conflict, and isn’t meant to. The challenge is structuring disagreement so decisions can still be made — and hold up to scrutiny — by clarifying tradeoffs, surfacing impacts, and narrowing options. Climate policy makes this especially clear; decisions about energy, land use, and infrastructure must move quickly while navigating disagreement about costs, risks, and local impacts.
Participation must reflect not just who shows up, but the full set of people affected, including those whose interests are less visible but equally consequential. Engagement can overrepresent highly organized or locally affected groups, even when decisions carry broader regional or national benefits. Designing participation to reflect those broader impacts — and the perspectives not in the room — helps avoid decisions that are responsive but not well-balanced.
The OMB memo advances a shift from participation as a single event (a hearing or listening session) to a practice agencies must design intentionally, tailor to context, and improve over time. The memo’s five principles — Purposeful, Respectful, Transparent and Accountable, Accessible, and Learning-Focused — offer a practical framework aligned with the decision, the stakes, and the people affected.
At their best, these principles help government:
- reach the right people (not just the easiest to reach)
- ask questions tied to real choices and constraints (not just general opinions)
- use input to shape decisions and follow-through (not just record it)
The goal isn’t consensus — it’s decisions that can move forward.Climate urgency makes getting this right non-negotiable. This framework is designed for exactly the conditions climate policy creates: high stakes, contested choices, deep skepticism, and no margin for processes that consume time without building trust.
Five Guiding Principles for Meaningful Engagement
Is it purposeful?
Purposeful engagement starts with clarity. What decision is being made? What is open to input at this stage? What is constrained by law, budget, or timing? Who is affected, and when can input still affect outcomes? It also means asking participants to respond to specific, decision-relevant questions. Engagement begins early enough for communities to help shape options, not simply react to them.
Why it matters
Without a clear purpose tied to a decision, engagement captures reactions to incomplete information or comes after key choices are already set. When choices are complex, participants may rely on partial or misleading assumptions rather than the factors shaping them. Framing questions around real constraints and tradeoffs produces more informed, actionable input. Inviting input outside an agency’s authority or capacity can also overwhelm staff and create unmet expectations.
What it looks like
- Engage before major choices are locked in.
- Present a limited set of realistic options (e.g., 2–3 alternatives).
- Ask targeted questions tied to specific considerations (e.g., cost, siting, mitigation, local impacts), not general preferences (e.g., “do you support this project?”).
- Define what input is in scope, and how out-of-scope input will be routed (e.g., partners, interagency coordination, future tracking).
Is it respectful?
Respectful engagement treats communities as knowledgeable partners and acknowledges the costs of participation. It reduces barriers where possible and ensures participation is worthwhile and relevant.
Why it matters
When engagement feels extractive, one-sided, or not worth the time required, participation drops and trust erodes, especially in communities already bearing environmental and infrastructure burdens. This affects not just who participates, but the relevance of the input received. People are more likely to engage — and stay engaged — when the process is clearly connected to decisions, provides enough context, and is worth their effort.
What it looks like
- Partner with trusted intermediaries to co-design or co-host engagement.
- Provide support (e.g., stipends, childcare, travel reimbursement) where feasible (see, for example, community compensation guidelines from the Colorado Department of Human Services and Washington State Office of Equity).
- Set clear norms for dialogue and how input will be documented.
- Train staff in facilitation, cultural competence, and navigating disagreement.
- Explain how input will be considered by decision-makers.
Is it transparent and accountable?
Transparent and accountable engagement sets clear expectations about what is being decided, how public input will be used, and how decisions will be communicated.
Why it matters
People don’t need to agree with outcomes to see them as legitimate, but they do need to understand how decisions were made. Transparency clarifies how input connects to decisions — including what can and cannot change — and helps maintain trust even when alignment is difficult. Accountability comes from closing the loop: showing what was heard, and what followed. Not all input will change outcomes, but its role should be visible.
What it looks like
- Communicate decision criteria, constraints, and timelines up front.
- Explain how input will be used before engagement begins.
- Share examples of how prior feedback influenced agency decisions or plans.
- After the engagement phase, show how input was considered by decision-makers (e.g., agency response to comments, draft revisions).
Is it accessible?
Accessible engagement removes practical barriers and proactively invites participation from those most affected but least likely to show up by default.
Why it matters
Barriers determine who participates and whose perspectives are heard. When participation depends on time, resources, technical fluency, or familiarity with government, input skews toward those advantages and may not reflect the full scope of public impacts. As a result, decisions rely on a narrower — and potentially distorted — set of inputs. Broadening access improves both representation and the quality of information decisions rely on.
What it looks like
- Offer multiple ways to participate (e.g., in-person, virtual, evenings, weekends) and provide input (e.g., written, audio, mapping tools).
- Use accessibility-by-default design principles (e.g., plain language, compatibility with assistive technologies, screen reader-friendly materials).
- Include stakeholders beyond those most visible or organized (e.g., future residents, regional beneficiaries).
- Conduct outreach through trusted community channels (e.g., local organizations, faith groups, libraries, community centers).
- Provide captioning, translation, interpretation, and other accessibility supports as standard practice — not only upon request.
Is it learning-focused?
Learning-focused engagement is iterative and adaptive. Agencies assess whether engagement is reaching the right people, producing helpful input, and informing decisions — and adjust accordingly.
Why it matters
Without iteration, engagement repeats the same gaps in participation and input. Learning in real time allows agencies to adjust how engagement is designed and delivered. It also prevents wasted effort by enabling course correction, saving resources and community goodwill — essential in fast-moving environmental and infrastructure contexts.
What it looks like
- Collect participant feedback (e.g., on clarity, accessibility, usefulness).
- Conduct internal debriefs to identify what worked and what didn’t.
- Monitor who is participating and who is missing.
- Adjust outreach, format, or framing mid-process.
- Use clear measures to assess the effectiveness of participation.
Is this engagement tied to a decision that is still open — and are participants being asked to respond to clearly defined choices, constraints, or tradeoffs?
Is this engagement worth people’s time — and are participants equipped to provide informed, relevant input?
Is it clear how input will be used — and how participants will see how it was considered?
Are we reaching and enabling participation beyond those most visible or organized — and who may still be missing?
Are we using what we learn to adjust this process in real time — and to improve future engagement?
Matching Methods to the Moment
No single method fits every situation. Effective participation matches the approach to the decision, based on who is affected, what is open to input, and what participation is feasible. Different stages of the policy lifecycle call for different forms of engagement:
- Early stages: define problems and surface lived experience.
- Mid-stage decisions: compare options and test tradeoffs.
- Implementation: troubleshoot and adapt.
The five principles set the standard for how to engage. The next step is choosing methods that apply them — fitting the decision, the audience, and the agency’s constraints (e.g., timeline, resources, legal requirements). Frameworks like the IAP2 Spectrum of Participation, the T.I.E.R.S. Public Engagement Framework, and the National Coalition for Dialogue and Deliberation’s Engagement Streams Framework can help avoid a common trap: defaulting to the same level or type of participation regardless of context.
Effective participation isn’t about asking more people more questions. It’s about selecting approaches that produce usable input.
That alignment also applies to who is included. Decisions with regional or national consequences require engagement that includes those who will benefit or bear indirect impacts. For example, housing or transmission projects often draw input primarily from current residents, even when benefits accrue to future residents or regional users.
This is not about giving any group veto power, but ensuring decision-making reflects the full distribution of impacts and interests.
Evidence from the Field
Well-designed participation isn’t just a process — it’s a governance tool. Examples from environmental and infrastructure policy show it can inform decisions, improve design, clarify contested evidence, and build the capacity for better engagement over time.
Participation that changes decisions
A 2025 study analyzing 108 Environmental Impact Statements under the National Environmental Policy Act (NEPA) found that public comments led to substantive changes in agency decisions in the majority of cases:
- 62% involved meaningful changes to decisions,
- 64% modified project alternatives,
- 42% changed mitigation plans, and
- when preferred alternatives shifted, agencies directly credited public input as the reason.
While the study did not assess outcome quality, longstanding NEPA success stories suggest these changes often strengthen project design, e.g., by identifying overlooked impacts, informing mitigation strategies, or incorporating local knowledge into technical analysis.
Place-sensitive design
Infrastructure decisions highlight the importance of place-sensitive engagement because impacts vary by location, history, and lived experience.
The Bipartisan Policy Center’s examination of a U.S. Department of Energy-funded carbon storage demonstration project in Illinois shows how early, sustained engagement helped build understanding and trust around geologic carbon storage. Engagement began years before site selection and relied on trusted local experts, multiple outreach strategies, and two-way communication to familiarize communities with the technology and its potential impacts. These efforts contributed to broad-based support and community willingness to host the project, illustrating how early engagement can shape perceptions of risk and benefit and improve the conditions under which projects move forward.
Similarly, analysis by Acadia Center and Clean Air Task Force found that opposition and delays were reduced, and public support for infrastructure grew, when clean energy planners took local siting and environmental concerns seriously and equipped communities to participate meaningfully.
These examples underscore an important balance. Place-sensitive engagement works best when local input is considered alongside broader system needs, so place-based concerns inform — but do not override — decisions with wider public benefits.
Joint fact-finding and shared inquiry
In disputes involving scientific uncertainty and contested values, joint fact-finding — where agencies, experts, and stakeholders collaboratively define questions, gather evidence, and interpret findings — produces more credible, usable information. Rather than positioning agencies and communities as adversaries, these approaches shift focus from competing claims to shared inquiry, helping participants develop a common understanding of facts and tradeoffs.
Environmental dispute cases show that joint fact-finding can narrow disagreements and reduce mistrust even when consensus is not possible. In practice, these processes help participants clarify what is known, what remains uncertain, and where value-based disagreements persist — allowing decisions to move forward on a more transparent and informed basis.
Environmental justice case studies documented by the U.S. Environmental Protection Agency (EPA) likewise illustrate how collaborative inquiry can enhance participation and buy-in from affected communities. In several cases, involving community members directly in data collection and interpretation improved the relevance of findings, increased confidence in the results, and fostered more constructive dialogue between agencies and communities — strengthening both the substance of decisions and their implementation.
Community-led research and data governance
Community-based participatory research offers another pathway to stronger decisions by changing who controls the production of knowledge. Analysis from the Brookings Institution shows that when communities help set research priorities and interpret findings, the results better reflect local context and needs. Traditional research models often reflect externally defined agendas that lack community-specific knowledge, limiting their usefulness for decision-making.
Community-led approaches, by contrast, redistribute control over research and data governance, enabling communities to shape how information is generated and used. While joint fact-finding focuses on how agencies, experts, and stakeholders collaboratively interpret evidence in decision-making contexts, community-led research changes who sets the agenda in the first place. In practice, this can produce more relevant inputs for policy and planning, strengthen the connection between data and lived experience, and support ongoing partnerships that extend beyond a single engagement process.
Evaluation
Effective engagement improves through feedback. For example, the U.S. Army Corps of Engineers’ evaluation of public involvement in flood risk management pilots found that engagement tied to clear decision points and structured activities (e.g., working groups, facilitated discussions) strengthened agency capacity for public involvement and improved two-way dialogue with communities. Participating staff reported that these efforts helped teams better understand community concerns, identify information gaps, and structure engagement more systematically.
Internal debriefs and participant feedback informed adjustments across project phases, helping teams refine outreach, coordination, and how input is organized and applied.
These findings spotlight another benefit of well-designed engagement: not just contributing to individual decisions, but building the knowledge, relationships, and processes that make more informed and collaborative decision-making possible.
What These Examples Show
Taken together, these examples point to a clear pattern: engagement works best when tied to decisions still being shaped and structured to produce usable input from those affected. In these conditions, participation does more than gather input — it improves decisions and delivery.
As noted earlier, participation doesn’t remove disagreement. It makes it more manageable by clarifying what is known and uncertain, surfacing tradeoffs, and reflecting a more balanced set of perspectives. This better equips decision-makers to explain and defend their choices.
Building Capacity to Deliver
Well-designed participation doesn’t substitute for agency capacity — it sharpens it, especially at the state and local levels where timelines are tight, staff are limited, and decisions are high stakes.
Participation is often treated as an added burden on already stretched institutions. But when targeted and structured, it helps agencies use existing capacity more effectively by identifying concerns early and reducing downstream conflict, redesign, and delay. This isn’t just an equity argument; it’s a speed and delivery argument.
What matters isn’t whether agencies “have capacity,” but whether participation is designed to support decisions that can be explained and sustained.
Strengthening State Capacity
Next, let’s look at the specific elements necessary to improve public participation.
Leadership and Governance
Why it matters
Engagement succeeds when it is treated as core governance, not a communications add-on. When leaders treat participation as part of decision-making, it affects how processes are designed, how staff are incentivized, and how tradeoffs are handled.
What it looks like
- Designate engagement leads to coordinate across programs and align input with decisions that cut across issues or policy areas.
- Embed clear ownership within program teams.
- Build engagement milestones into project timelines.
- Have senior leaders review engagement summaries alongside legal, technical, and budget analyses.
- Reflect participation goals in agency strategic plans, implementation plans, and performance reviews.
Skills and Culture
Why it matters
Engagement failures are often organizational, not technical. Without the right skills and norms, staff may struggle to use public input or navigate conflict, and even well-intentioned engagement can break down.
What it looks like
- Train staff to interpret and weigh public input alongside technical and operational constraints.
- Synthesize input into themes and areas of agreement or tension so it can be compared and shared without revisiting individual comments.
- Design engagement plans across functions (policy, legal, technical, communications).
- Set expectations that engagement is part of policy development and implementation, not a parallel process.
- Reinforce these norms through staffing, timelines, and accountability.
Tools and Resources
Why it matters
Without the right tools and resources, engagement can generate more input than agencies can realistically analyze or respond to. The issue isn’t just volume, but whether input can be organized, interpreted, and applied to decisions.
What it looks like
- Use practical checklists and templates for planning, documentation, and follow-up.
- Use formats that produce comparable, decision-relevant input (e.g., facilitated discussions, guided prompts, prioritization exercises).
- Use analysis approaches that account for different types of input and perspectives (e.g., written comments vs. oral input, surveys vs. community discussions), not just the easiest responses to process.
- Establish pre-approved contracting mechanisms for facilitators, translators, and interpreters.
- Track commitments and follow-through internally (e.g., via dashboards).
Learning and Improvement Systems
Why it matters
Adaptation requires more than intent. Learning at the project level isn’t sufficient — without shared systems, agencies tend to apply the same approaches across teams and over time, regardless of effectiveness.
What it looks like
- Standardize internal reporting that connects public input to decisions and next steps (e.g., “what we heard / what we’re doing” summaries).
- Establish criteria for adjusting outreach or formats based on participation patterns (e.g., extend timelines if turnout is low, add targeted outreach to missing groups).
- Create shared repositories so lessons learned carry across teams and inform future projects.
- Incorporate outcome checks over time (e.g., whether engagement reduced conflict or improved implementation).
Strengthening Public Capacity
This challenge extends beyond agencies themselves. Participation design should account for both agency capacity and who can realistically participate. When engagement skews toward people who face fewer barriers to participation, it raises equity concerns and weakens the quality of information and problem-solving.
Reducing Participation Barriers
Why it matters
Reducing barriers isn’t about paying for feedback. It’s about making participation feasible, informed, and reflective of those most affected, including those who will experience the long-term impacts.
What it looks like
- Provide context in advance so participants can engage without technical expertise.
- Provide information in multiple languages and accessible formats.
- Share clear examples of useful input (e.g., specific impacts, leading practices).
- Present side-by-side comparisons of relevant options and tradeoffs (e.g., anticipated impacts, costs, timelines).
- Offer opportunities for the public to ask clarifying questions before providing input (e.g., Q&A sessions, office hours).
Equipping Communities to Engage
Why it matters
Meaningful engagement often requires skills, time, and capacity that some communities — especially smaller or under-resourced ones — may not have. Without intentional outreach and resourcing, agencies hear repeatedly from the same well-resourced groups. Just as important, without support for organized participation, engagement struggles to translate into representative voice, particularly for communities historically marginalized by race or class.
What it looks like:
- Provide practical tools to support effective participation (e.g., how-to videos, comment templates, guides to agency processes).
- Share meeting materials early so communities have time to review, discuss, and respond.
- Provide small grants or stipends so trusted partners can convene discussions, and synthesize input, and support representative participation.
- Designate clear agency points of contact to help communities navigate participation processes.
Developing Long-Term Relationships
Why it matters
Engagement is faster and more collaborative when relationships already exist. When agencies engage only during moments of controversy, participation is more likely to feel reactive and transactional.
What it looks like
- Maintain engagement beyond one-off interactions, including through light-touch ways (e.g., periodic newsletters, community listservs).
- Maintain continuity in points of contact through clear handoffs and shared records so relationships and institutional knowledge persist as staff change.
- Communicate regularly and follow through outside active decision processes.
- Acknowledge past harms or broken trust.
- Return to communities after implementation to share outcomes.
Return on Investment
Trust, as this paper has argued, is the load-bearing wall. Investing in state and public capacity builds it — so participation helps decisions progress rather than stall. Well-designed engagement enables agencies to use input and respond credibly. When communities can participate fully, decisions are better grounded, easier to explain, and more likely to hold.
Looking Ahead
As engagement tools evolve, the same principles apply. Digital participation, civic technology, and AI-assisted analysis are already being used to help governments reach more people and make sense of large volumes of input. But without deliberate design, they risk introducing new exclusions and harms, such as unequal access, privacy concerns, and bias.
Internationally, several examples illustrate both the promise and the tradeoffs. France used AI tools to analyze millions of contributions submitted during the Grand Débat National, grouping input by theme to make citizen insights more accessible to policymakers; the process highlighted tensions between scale and nuance, and raised questions about how input is aggregated and whose perspectives are preserved. The UK government uses an AI consultation analysis tool to analyze consultation responses at scale, reducing manual work and improving the speed and consistency of analysis. Its evaluation documents practical challenges, including the need for human review and concerns regarding bias, accuracy across groups, public trust, and transparency in interpretation.
These examples point to real potential: making participation more scalable, accessible, and usable in decision-making. But they also reinforce the same lesson as the rest of this paper — the impact of these tools depends less on the technology itself than on how they are designed and governed.
The path forward is not to adopt emerging tools wholesale, but to hold them to the same standards.
- Purposeful: Use tools to help answer specific questions or synthesize input, not simply because they are new or project “modernization.”
- Respectful: Handle community data, stories, and participation with care, including clear consent and appropriate privacy protections, especially when considering AI tools beholden to terms of use agreements.
- Transparent and Accountable: Explain how input is collected, analyzed, and used, especially when automation or AI is involved.
- Accessible: Design for inclusion rather than assume technology access or digital fluency.
- Learning-Focused: Assess whether tools improve engagement and decision-making, and adjust or retire tools that do not.
Used this way, technology doesn’t replace judgment or governance; it strengthens both.
As agencies adopt new tools, they can also build on existing human infrastructure — such as promotores and other community-based messengers — that has long been used in public health and service delivery to support outreach, build trust, and connect agencies with communities.
When participation is designed to clarify tradeoffs, surface real impacts, and support accountable decisions, it becomes what democratic governance needs most right now, and what this collection argues is still within reach: a government that listens well enough to be worth believing in.
Strengthening the Federal Cycle of Learning and Adaptation by Closing the Loops
The federal government has a feedback-loop problem.
Regularly generated information, including evidence, performance information, and qualitative insights from implementation, too often fails to shape decisions. Evidence may be reviewed without changing priorities; performance data may be tracked without clarifying what it informs, and implementation feedback may reach leadership without surfacing what works for whom and why, or suggesting next steps. The components of a cyclical learning system linking priorities, questions, evidence, decisions, and implementation information exist in theory and on paper, but the connective tissue that turns all of these components into a functioning cycle of learning and adjustment is lacking. Information and artifacts alone don’t necessarily facilitate learning and adaptation; strengthening federal feedback loops requires embedding translation and use into decision-making from the start.
This memo is not a case for new infrastructure. The Evidence Act, learning agendas, evaluation plans, performance frameworks, and customer experience authorities already exist; what they do not yet add up to is a learning system. The translation this memo proposes is turning the infrastructure we have into the learning system we need, and it’s addressed to federal program leaders, policy officials, evaluation and evidence staff, performance officers, and strategic planning teams who already sit inside it and are best positioned to make it function as intended.
Challenge and Opportunity
The federal government already operates within a broad cycle of goal-setting, evidence generation, performance review, implementation, and reporting. On paper and in principle, this cycle should allow for learning, adjustment, and improvement to federal programs over time. In practice, however, agencies vary in how consistently they translate such information into planning, decision-making, or course-correction. Federal agencies have made progress in building and using evidence, but translating that information into timely operational or policy revisions remains uneven.
The core problem isn’t production; it’s translation, and the translation failure shows up as “so what” gaps on both sides of the information pipeline. On the input side, receivers of information are often left asking what they’re supposed to do, and on the output side, a second question appears – “is it my job to act on this, and if so, how?”. Research findings are often too slow, too caveated, or too disconnected from immediate policy and management questions. Performance data may show quantitative changes in outputs, costs, or enrollment without revealing the mechanisms behind them or the practical implications for implementation, or cueing the design apparatus that could apply these insights. Feedback from frontline service providers and affected users might reach leadership mainly through quantitative indicators, dashboards, or status updates, which don’t always capture lived experience, causal explanation, or informed suggestions for course correction. Without named owners and defined next steps, even the most actionable information tends to circulate rather than convert.
Three gaps sit behind this pattern. First; a context gap – decision-makers often lack the full picture, because qualitative indicators and customer experience research arrive separately, or later than quantitative evidence, leaving them with only a partial view of what’s working well or driving implementation problems. Second, an action gap; even with a complete view of the picture, it’s not always obvious which lever applies, on what timeline, or with what tradeoff. Third, an ownership gap; it’s often unclear who is responsible for translating any given signal into a decision, and this ambiguity means that insights can be observed without being acted on. Together, these three gaps leave evidence and feedback insufficiently integrated into decision-making routines.
The problem is also structural; decision-makers face turnover, competing priorities, time limits, and management pressures, and thus, evidence needs a more robust pathway. Devoid of clear translation, trusted messengers, and defined or mandated use points, even the most relevant information can be too late, too ill-timed, or too jargon-heavy to influence decisions, resulting in missed adaptation opportunities.
The federal government doesn’t need an entirely new learning architecture. It needs to make the one it already has more usable. Agencies can do this in a few ways. First, by building stronger translation functions by creating space for “knowledge brokers” (people or teams whose core function is to translate evidence into decision-relevant language and maintain the required relationships that make the translation trusted). Second, by incorporating the use of evidence, performance, and implementation feedback into policy and program work from the start. Third, by creating better pathways for implementation and lived-experience feedback to reach leadership in ways that resonate with them and support action.
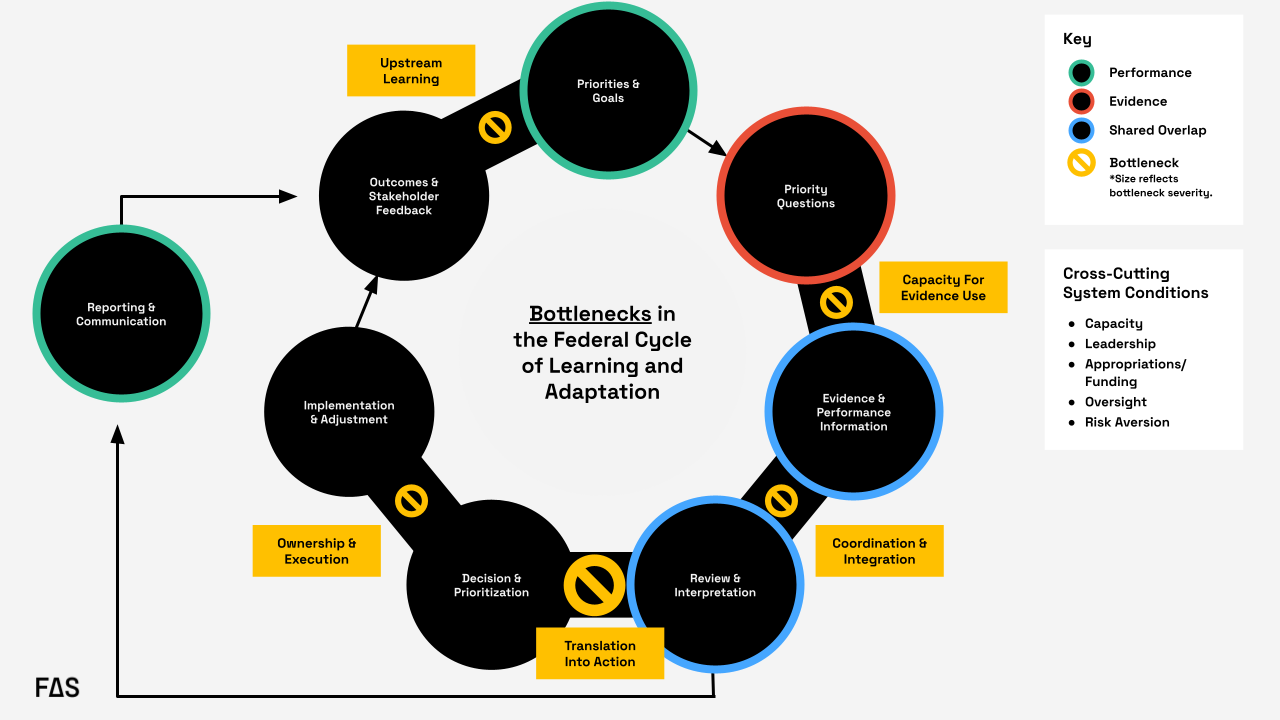
Formal federal guidance envisions a closed “loop” linking goals, priority questions, evidence, and performance information, along with review, decision-making, implementation, reporting, and feedback. In practice, the loop often degrades at key handoffs: evidence-use capacity, coordination and integration, translation into action, ownership and execution, and upstream learning from outcomes. The most significant recurring bottleneck occurs during the transition from review and interpretation to decision and prioritization, where information is generated and reviewed but does not reliably translate into action.
Plan of Action
We need to shift from a system that collects information to a system that uses it.
Agencies should create or strengthen embedded translation functions that connect evidence, performance information, implementation experience, and policy levers at the moment decisions are being made. The key is to move from a dissemination model to a utilization model. Instead of “produce, disseminate, and hope for uptake”, agencies should do the following:
Recommendation 1. Designate a knowledge broker to facilitate regular decision briefs…
or routines that create structured opportunities to clarify what’s known, what’s uncertain, and what actions are available – beginning with a defined set of high priority issues rather than every single decision the agency needs to make.
This recommendation targets the translation into action bottleneck in Figure 1; the handoff from review and interpretation to decision and prioritization, which can be considered the most significant recurring point of failure in the federal cycle of learning and adaptation. In practice, this means assigning this function to a role or small team housed in an existing performance, evaluation, strategy, or program office and requiring that group to support recurring decision points with short decision briefs. Those briefs should identify the decision, synthesize relevant evidence, performance trends, and implementation feedback, and specify available actions, tradeoffs, and owners.
For example, in the rollout of the FAFSA Simplification Act, a knowledge broker tied specifically to this initiative could have translated readiness indicators, beneficiary feedback, and information from financial aid administrators into decision-ready synthesis for the officials with decision authority who were attempting to course correct in real time. Instead, significant delays turned the rollout into a high-profile implementation issue.
Crucially, this function should start narrow. Rather than positioning a knowledge broker as an all-purpose translator for an agency’s full decision load, the initial portfolio could be scoped to a small set of priority issues. Starting narrow lets the broker establish credibility and relationships that make translation trusted and refine what the routine and explicit outputs are before it scales – the portfolio can scale later. This gives agencies a defined mechanism for turning reviewed information into decisions rather than leaving that handoff informal. Within the federal government, the Office of Evaluation Sciences has modeled how an embedded team of evidence translators can work alongside program offices rather than from a silo.
Recommendation 2. Start policy initiatives and evaluation planning with a real question…
or decision, identify the user, specify the lever, and clarify – in advance – what different findings would imply. Incorporating this thinking upstream changes the role of evidence from a retrospective input to an operational tool.
This recommendation mainly addresses the translation into action bottleneck, and secondarily, the evidence-use capacity bottleneck. Agencies can operationalize this by building decision framing into existing learning agenda and evaluation planning processes, both of which are already required under the Evidence Act. Before an evidence product is commissioned or a performance indicator is selected, program offices can be required to answer four questions on the record: What decision is this for? Who will use it? What lever would change as a result? What finding would lead to what action?
In a hypothetical example, say that USDA’s Food and Nutrition Service (FNS) wants to commission a new evaluation or analysis regarding SNAP redetermination churn (the pattern of households losing SNAP benefits at recertification and then re-enrolling, often for procedural reasons rather than eligibility). The four questions noted above can be answered on the record before the work begins. The decision is whether to issue new guidance to state agencies on recertification practices and what that guidance should encourage. The user is the FNS administrator and the relevant policy office, with state level SNAP directors as the implementing audience. The lever is subregulatory guidance. The early thinking regarding mapping findings to actions would specify, in advance, which patterns or insights would trigger which associated response. This way, when the findings arrive, USDA wouldn’t be starting from scratch with the “what do we do with this” question; the decision architecture would already be in place.
Pre-specifying these conditions in a short decision-framing memo that travels with the work turns evidence from a retrospective deliverable into a tool scoped to a specific decision or policy window. The same logic extends to decision memo templates themselves, which can include standing prompts such as “what evidence informed this decision?” and “how will we learn about this in real time?”, so that utilization is built in.
Recommendation 3. Create pathways for easier access to mixed methods evidence and insights from lived experience.
Recommendation 3 targets the coordination and integration, and upstream learning bottlenecks; the gap where evaluation, performance, administrative, qualitative, and customer experience data move at different speeds, live in different places, and reach decision-makers as parallel streams. Insights from lived experience – what programs actually look and feel like to the people using them – are particularly likely to be separated from information that reaches leadership, arriving as anecdotes, if they arrive at all. Because these problems are distinct; the recommendations can be broken down and addressed at the agency level.
Recommendation 4. Standardize decision-ready formats that consolidate quantitative and qualitative evidence.
Agencies should build standardized decision templates and briefs that present quantitative indicator-level data alongside narrative summaries of lived experience and implementation conditions, so decision-makers aren’t expected to synthesize across disparate sources on their own timelines. The resulting artifacts should be tied to recurring decision moments (budgeting, guidance revisions, program reauthorization) so that they can be used in real time.
In the FAFSA Simplification Act rollout, the Department of Education leadership faced this problem: application data, technical readiness indicators, information from financial aid administrators, and user feedback existed separately, and moved at different speeds. A standardized decision-ready format could have pulled those streams together; pairing completion trend data with brief narratives or exemplary quotes regarding what applicants and financial aid offices were actually encountering, rather than leaving leaders alone to assemble the picture in real time
Recommendation 5. Actively use existing general clearance mechanisms for rapid qualitative and user experience research.
Agencies should make use of standing generic clearance mechanisms that allow them to fast-track small qualitative and user experience studies (for example, up to 100 respondents, completed within a fixed time window) when unexpected findings need rapid explanation. This would allow for the ability to run a tightly scoped evaluation in weeks rather than months, which is the operational timescale at which decisions frequently move. Without it, the qualitative evidence needed to explain any type of performance anomaly often arrives after the decision or policy window has closed.
For SNAP redetermination churn, this would let FNS turn around a short, scoped evaluation of why participants are dropping off at a specific step in the recertification cycle in weeks rather than months. The insights could then inform the next round of guidance rather than coming in after the fact.
Recommendation 6. Build customer-first indicators built into existing federal reporting requirements.
Beneficiary and frontline experience should become part of the evidence base by default rather than by exception. Most federal programs already have reporting infrastructure, and layering in a modest set of customer-first indicators that use the existing infrastructure rather than building new information collection requirements ensures that user perspectives are consistently available as routine inputs.
Within the federal government, the customer experience and life experience work coordinated through OMB and performance.gov has demonstrated that lived experience can be collected and used at scale within existing authorities, which can be considered a foundation to build from rather than reinvent. At the state level,Minnesota’s Story Collective, housed within Minnesota Management and Budget (MN MMB), pairs administrative and performance data with qualitative, lived-experience narratives to give decision-makers a richer view of what their programs are actually producing.
These recommendations also address a common weakness in the federal system: evidence and performance information sit within the same broad ecosystem but move at different speeds, use different tools, and often reach different audiences. This is all the more reason to create a translation layer that can synthesize across them. Agencies need staff and routines that can connect evaluation, administrative data, performance indicators, qualitative input, and implementation realities into decision-relevant guidance. Without that connective tissue, agencies are left with parallel streams of information that don’t consistently converge at the point where action occurs.
Conclusion
The federal government already generates a great deal of information about what it’s doing and how it’s performing, but information isn’t the same as learning, and learning isn’t the same as adaptation. The gap between them is where the “so what” goes unanswered, and where the federal feedback loop breaks down.
Closing these loops doesn’t require new infrastructure or authority. It requires three shifts in how the existing system is used: designating knowledge brokers to carry translation across the handoff from review to decision-making, building decision framing into policy and evaluation work from the start so that evidence is scoped to the decisions it’s meant to inform, and creating pathways that move mixed-methods and lived experience into decision-makers’ hands in formats and timeframes that match how decisions actually happen in the federal environment. Whether the question is about USDA responding to SNAP redetermination churn or the Department of Education learning from an application rollout in real time, the underlying pattern is the same: the signals exist, but the translation that turns signals into actionable insights doesn’t reliably happen.
If the government wants a system of learning and adaptation that improves results in real time, it has to treat translation, utilization, and adaptation as core functions of governance rather than as afterthoughts.
Why Interagency Policy Coordination Efforts Frequently Fail (And How To Fix Them)
One of the great innovations of the American experiment is the federation of the organization of government at scale. For all the advantages to science and technology governance that federalism has promoted, the decentralization of responsibilities within the federal government makes speaking with one voice difficult. While differences between government agencies has been a source of strength for the U.S. federal research and development (R&D) ecosystem, those differences can also manifest as tribalism in the governance of programs administered and regulations promulgated by those agencies.
We firmly believe that it’s possible to “stack the deck” in such a way that produces better governing outcomes with stronger public impact while maintaining the strength inherent in interagency deliberative processes. To do that, this memo hopes to demystify the way that decisions are made inside agency bodies while providing a roadmap that can help decisionmakers and their advisors achieve those better outcomes. Our recommendations encourage senior policymakers and policy influencers, alike, to maintain a strong teleological focus, to embrace economic impact assessment, and adequate resourcing and troubleshooting gumption to deliver tangible policy outcomes.
Differences in R&D priority implementation between agencies can be subtle, such as a discrepancy in legal interpretation over a federal statute, or they can be overt, such as one agency taking a position seemingly at odds with another, or procedural such as when two agencies require redundant paperwork to collect information. This lack of cohesion can ultimately lead to significantly increased administrative burden for stakeholders, slower deployment of Administration priorities, and increased costs for the R&D ecosystem. To address this, R&D stakeholders frequently ask for more harmonized rulemaking between agencies. Unfortunately, the incentive structures supporting institutional autonomy frustrate harmonization and simplification efforts, further slowing the work of the government and (in some cases) magnifying the administrative burden experienced by stakeholder organizations.
Challenge and Opportunity
Coordination among federal science agencies is essential to ensure government-wide alignment on R&D investment priorities and policy agendas. However, the federal R&D enterprise suffers from some of the most egregious siloization of process and procedure in the administrative state.
To give one specific example, non-governmental space activities are covered by multiple agencies, with the Federal Aviation Administration covering launch and reentry, the Federal Communications Commission and Department of Commerce covering spectrum-related issues, the Office of Space Commerce covering remote sensing technologies, and the Federal Communications Commission (strangely) covering orbital debris. If a company wishes to engage in remote sensing activities, they need to engage each of these licensing processes, separately, which may operate on different timelines and with different levels of priority. Both the Trump administration’s executive order on commercial spaceflight and the Biden-Harris administration’s novel space activities proposal maintain the separation of licensing processes between crewed and uncrewed space activities, protecting the equities of both the Department of Transportation and Department of Commerce rather than resulting in regulatory rationalization and placing them at odds with with Congress’ proposals.
Disharmony in the regulatory system may result in differentiation of federal grant, contract, and cooperative agreement requirements that organizations need to navigate; oftentimes requiring multiple and substantially different layers of expertise and government-service provider relationships that organizations have to manage. When the government does coordinate, interagency bodies frequently develop recommendations that maximize the flexibility of the government institutions, but may avoid addressing fundamental ethical issues or the concerns of impacted communities.
To address these complaints and ensure alignment, both formal and informal interagency deliberative bodies are often formed within the Executive Branch. These may take the form of committees, subcommittees, and working groups of Congressionally mandated interagency fora, such as the National Science and Technology Council or the Interagency Council on Statistical Policy. Or, they may arise from the convening power of components in the Executive Office of the President, in what are known as, policy coordination committees in the current administration.
Less often, they form organically through recognition for the need of coordination by the agencies themselves, such as Commerce, Energy, NASA, Defense Information Managers Group (CENDI). These bodies, and other ad-hoc working groups, are almost always formed by enterprising individuals in more junior government roles when there are clear mission requirements which need to be addressed. Such groups can be incredibly effective, especially if organically-formed groups are well-acquainted with legal and funding constraints, but can also be relatively limited in overall ambition.
To navigate thorny issues and resolve interagency disagreements, senior staff, members of Congress, and even members of federal advisory committees often turn to these bodies, or seek their creation. Take the recent example of the National Academy of Science, Engineering and Medicine report “Simplifying Research Regulations and Policies”, whose recommendations focus primarily on increasing regulatory harmony between government agencies rather than actually removing or changing government requirements.
Incentive Structures Favor the Status Quo
Agencies have enormous incentives to protect their turf – particularly their legal authorities – under these coordination bodies. Given the uncertainty of funding and legislation, and the risk that dedicating work toward a particular task might result in a reallocation of resources from something that agencies are already assigned to do, the most common output created by these interagency bodies is a line of best fit that deftly navigates the specific interests of each federal agency, sometimes with compromises that water down Executive or Legislative intent.
Worse, agencies in question might not have available time or resources for officers to dedicate the type of high-level attention needed to actually implementing an ideal solution. As a result, the chair for these committees often takes the lead in developing a work plan, which is usually heavily-shaped by individual experiences within a single government agency or even single industry. If there is significant political support or pressure for the interagency organization to produce a product, like a proposed regulation or report, other government departments and agencies might not even weigh in substantively to maintain plausible deniability. Instead, such agencies may seek to minimize the number of new requirements or dilute requirements to avoid affecting agency operations.
In an extreme example of the consequences of understaffing an interagency coordination body while concentrating one agency’s perspective in a single individual with little actual authority, the Department of Health and Human Services (HHS) unwound nearly eight years of bilateral negotiations between the United States and the European Union during the Biden administration because their subject matter expert unexpectedly and unfortunately passed away during the negotiation’s final stages. Ironically, this was a topic of great HHS interest – improving the ability for US and EU researchers to share human health-related data across the Atlantic. HHS’ failure to appoint a new champion effectively ended the negotiation, fueled by the agency’s claims that appropriate vetting of the final policy could not be prioritized given staffing demands and a lack of expertise. The impacts of this non-decision were not localized to government laboratories, but also stakeholders across industry and the biomedical research ecosystem.
The products of these coordination committees generally favor the experience of the chair, moderated primarily by the government’s ability to respond to a new (usually unfunded) demand within existing resources. This seldom answers the mail requested by the Executive Office of the President, Congress, or the recommendations offered by outside advisory groups, and can place costly new requirements on the rest of the ecosystem.
Thou Shalt Not Suggest Future Financial Commitments are Needed
One of the perennial challenges that interagency coordination groups face is the fact that recommendations for action may conflict with the annual President’s Budget Discretionary Request (PBR). This is based on the expectation that the Executive Branch speaks on budget issues with one voice. Likewise, the Anti-Lobbying Act is generally understood within the Executive Branch to prohibit agencies from seeking additional funding or appropriations. Responses to Congressional requests are often vetted by the Office of Management and Budget.
This is complicated by the fact that when Congress asks an agency for a proposal on how to do something, there is a general expectation that the Executive Branch provide a clear and comprehensive answer that will allow legislators to act expeditiously on resolving the problem, particularly with respect to how Congress can help. This also happens when agencies respond to crises, when political appointees seek to open new lines of effort, or when international partners come to the U.S. government with a proposal that is generally favorable to the President. Even if the President favors acting on the proposal, it can be difficult for agencies to support action that is not accounted for in the PBR.
Because these requests generally lack funding information that would place the recommendations outside the scope of the PBR, there are four possible outcomes:
The interagency group will often:
- Develop a recommendation that places resource burdens on entities that are not a part of the federal government (e.g., through the establishment of additional unfunded mandates placed on organizations seeking federal grants, contracts, or cooperative agreements);
- Provide with a “zero cost solution” that minimizes the resource expenditure required of the government institution but may not actually solve the problem;
- Remain silent or blame Congress (or the White House Office of Management and Budget (OMB)) for not providing sufficient funding to achieve a particular objective, sometimes forcing committee staff to seek outside advice on what implementing recommendations might cost. Such efforts could represent a de facto information collection, increasing the burden placed on the affected stakeholders; or
- Fail to answer the request after the interagency coordination effort breaks down or loses its champion.
Obviously, these results inherently mask the true cost of the government’s activities and minimizes the work undertaken by the executive agencies. One specific example that we encountered in our time in government involved a suggestion that the interagency group explicitly state that individuals stranded in space or on other planets on commercial space missions would not be rescued because the government had not and would not request funding for such an eventuality. The interagency group ended up not releasing their product.
Former staff who served in the current administration have offered their solution to the coordination issue: only request input from agencies when their perceived equities are in play. The problem with such an assumption is that other government agencies frequently have equities that are not immediately visible to the Executive Office of the President or other government bodies, creating a probability that the exclusion of that agency could result in them becoming a process spoiler sometime in the future. Sometimes agencies use this as a tactic to kill or stall an initiative by intentionally falling to staff participation on an interagency body and then complaining at the 11th hour that they weren’t consulted on the group’s policy product. When rulemakings are in play, the interagency deliberative processes defined by Executive Order 12866 are supposed to be followed to prevent this type of skullduggery. However, even then OMB’s Office of Information and Regulatory Affairs (OIRA), which coordinates and enforces the process, often provides agencies (and even external organizations) with a great deal of latitude to effect their delay tactics.
Agency Staff Feels the Need to Protect their Decisionmaking Authorities
In the words of one of our former coworkers, “agencies will do anything to protect their ability to make an independent decision.” Coordination bodies frequently are expected to set priorities that could tie the hands of a government agency (or the Presidency) at a future date. This is particularly acute in science, when the funding cycle and normal life of a scientific project usually doesn’t align with the political calendars of Congress or the Presidency. As a result, agency staff and their political leadership are disincentivized to make recommendations that would limit their ability to make a different decision at some point in the future, or encouraged to non-concur to protect the ability of current staff to interject. Some agencies, particularly the Department of Health and Human Services and the Department of Defense, are particularly aggressive in using these tactics given their substantial resources and long-mission timelines which may be vulnerable to future political disruption.
Ultimately, the aspiration for a unified federal vision often founders on the jagged rocks of agency tribalism. When agencies prioritize the preservation of their legal, administrative, or cultural turf and discretionary autonomy over the broader implementation of top-down mandates, interagency coordination transforms from a tool of progress into a performative exercise in risk mitigation. This protectionism creates a gravity well of inertia, where the resulting vector rarely points toward innovation or rationalization, but instead toward a diluted status quo that safeguards agency equities at the expense of (potentially) good governance. Until the incentive structures of the administrative state are realigned to reward cross-cutting outcomes over parochial independence, even the most robust top-level policies will continue to be slowly hollowed out by the very departments tasked with their implementation.
Plan of Action
Governing is hard. It forces organizations to make difficult decisions, accounting for tradeoffs and mitigating internal and external organizational risks. When Congress or the President ask an organization to provide recommendations on how to solve a problem, ideally the result should include a credible solution. Here, we recommend a path forward that protects the benefits of a federated system (strategic redundancy, checks-and-balances, etc) while minimizing tribalism that works against efficiencies of interagency coordination and implementation of policies.
Recommendation 1. Formal advisory committees to the government should be limited from deferring actions within their scope to interagency deliberative bodies.
This could be achieved simply through a White House-level policy memorandum, though it could also be accomplished in agency practice through charge letters to advisory groups, updates to policy for the National Academies and DC think tanks, or instructions in Congressional charging legislation.
When advisory groups like the National Academies of Sciences, Engineering, and Medicine produce recommendations or reports for the executive branch, the advisory committees formed to do the work will frequently recommend that an interagency body coordinate, develop strategies, or produce recommendations aimed at resolving a high level policy objective. For instance, the 2022 report “Protecting U.S. Technological Advantage” produced four recommendations that requested the creation of six new government processes to create new definitions, assess vulnerabilities, identify essential elements, develop a national strategy, and develop a new policy framework. By doing so, the advisory committee delegates its responsibility to recommend action and sets up a cascade of additional administrative burden that flows to the federal agencies (if they accept the challenge in the first place).
In the case of the “Protecting U.S. Technological Advantage” report, the end product implicated multiple government organizations that were not involved in the creation of the report (including numerous White House components) making it less likely that the recommendations would be adopted. Recommendations focusing on the outcome members of the Committee wanted from the government, in addition to the authorities or processes required to effectuate the outcome, would have made it easier for individuals in the government to support the recommendations and seek Congressional support.
Similarly, recommendations that suggest stronger interagency coordination, for instance to harmonize interagency policy and practice to reduce administrative burden, tend to ignore the factors cited earlier about the various factors that pull against successful policy coordination efforts. Instead, recommendations that weigh the tradeoffs associated with specific policy options and suggest answers to hard decisions are more likely to provide the information sought by policymakers and lawmakers, alike.
Decadal surveys, on the other hand, tend to provide science agencies and congress with relatively straightforward lists of priorities under different budget scenarios that are easily understood by budget officials and appropriators. While these recommendations are seldom followed in total by government agencies, they are proven delivery tools for program and policy implementation.
The government already takes similar measures in multilateral international activities. “Instruction cables” provided to members of U.S. delegations to international working groups contain express instructions to limit the creation of new working groups or bodies under the international organization to limit the proliferation of such mechanisms in international fora. Charge letters from agencies, language in Congressional authorizing legislation for National Academies studies, and other directives limiting the proliferation of new working groups and committees could include similar language.
Recommendation 2. Recommendations from interagency bodies that invoke new agency or impacted entity actions should include economic impact assessments.
Congress should require that interagency bodies identify and publicly disclose the costs to implement the group’s recommendations, including any expected costs to non-government organizations when those recommendations involve any changes to agency or impacted entity behavior. Interagency reports often set ambitious goals without accounting for the unfunded mandates they create for agencies and the private sector. The National Science and Technology Council’s October 2022 National Strategy for Advanced Manufacturing outlined sweeping goals for industrial decarbonization and supply chain restructuring, yet it lacked a granular economic impact assessment regarding the capital expenditures required by small to medium enterprises to comply with suggested standards. Without this data, recommendations risk becoming aspirational documents rather than actionable blueprints because the entities responsible for execution are left to discover the true costs only after the policy momentum has already shifted. Moreover, this leads to further fragmentation, redundancy, and increased burden across regulated sectors as any resulting rulemaking acting on these recommendations would have to engage its own separate cost analysis.
The consequences of omitting economic analysis are evident in recommendations regarding cybersecurity protocols often generated by Policy Coordination Committees. For example, the interagency implementation plans following Executive Order 14306 on Sustaining Select Efforts To Strengthen the Nation’s Cybersecurity often encourage agencies to adopt stringent data sharing frameworks without quantifying the compliance drag on regulated entities in the energy and telecommunications sectors. When recommendations do not account for the labor hours or hardware upgrades necessary for implementation, they can lead to significant market friction and cost surprise. By requiring an assessment at the outset, Congress can prevent the adoption of technically sound, but economically disruptive, policies that may stifle the innovation they aim to protect. Congress did enact the Unfunded Mandates Reform Act of 1995 to limit the impact of these types of interventions, but the application of the act is limited to state, local, and tribal governments (as opposed to industry or academic sectors).
Conversely, when interagency bodies integrate economic assessments, the resulting policy is more resilient and credible. The Interagency Working Group (IWG) on Social Cost of Greenhouse Gases serves as a primary example of a body that centers its output on economic modeling, as seen in its February 2021 Technical Support Document: Social Cost of Carbon, Methane, and Nitrous Oxide. By providing a quantified value for the social cost of carbon, the IWG ensured that any subsequent agency actions are grounded in a rigorous benefit-cost framework. Similarly, certain Federal Advisory Committee Act committees such as those advising the Department of Transportation frequently include economic subcommittees to vet recommendations. These examples show that when the economic price tag is transparent, stakeholders can provide more meaningful feedback and agencies can prioritize actions that offer the highest return on investment.Integrating economic impact assessments into the interagency process aligns these high-level recommendations with the gold standard of federal regulatory governance found in Executive Order 12866 and the Office of Information and Regulatory Affairs review process. Currently, a significant gap exists where a policy may be developed in an interagency report without the rigorous economic and regulatory impact analyses required during rulemaking. By requiring these assessments earlier in the deliberative stage, this recommendation ensures that the logic of balancing societal benefits against compliance costs is present from the moment of inception. This prevents regulatory path dependency where an agency feels compelled to codify a recommendation that has not yet faced the scrutiny of economic trade-offs, which enhances both transparency and fiscal responsibility.
Recommendation 3. Congress should fund its reporting demands on the interagency, particularly to ensure adequate staffing and to avoid diverting resources away from mission-focused activities.
Congress should appropriate resources when it makes reporting mandates, including to already-established reporting programs and interagency deliberative bodies. This would ensure the project actually happens and can be carried through to completion. This also frequently means that staff are diverted from their core duties to produce these reports, creating an additional drain on agency resources.
This challenge is particularly acute for White House components like OSTP, which are not budgeted to fulfill the number of recommendations and reporting obligations placed on the organization by Congress. Such reports are often left to the initiative of short-term detailees and Intergovernmental Personnel Act (IPA) appointees who may be unable to thoroughly answer the reporting requirements relevant to their portfolio while they are also satisfying Administration demands. As a result, reports connected to immediate political demand signals are generally prioritized over longer-term planning exercises and strategic assessments, leaving much work – including work tasked by Congress by law – frequently incomplete.
Recommendation 4. “Center of government” organizations, like OMB, OSTP, and the National Security Council (NSC) should actively seek and provide funding and direction to support Administration goals and objectives.
Such efforts should be supported by champions, both at the senior level in the center of government organization as well as champions within the interagency who have the authority to clear bottlenecks and sufficient top cover to seek necessary funding and other resources.
It is an oft-stated maxim in government “show me your budget and I’ll show you your strategy.” When agencies are asked to do something based on a top down directive, political resources should be aligned with ensuring that the effort succeeds. Frequently this is not the case.
In international affairs, the visit of a high-level foreign official will frequently trigger requests, most often from the NSC and the U.S. Ambassador to a country for projects or programs that can be offered to demonstrate to the foreign partner the value of the bilateral relationship between the two countries. There is just one problem–with the exception of defense and some security programs, government agencies almost never have dedicated resources to support such requests. The situation becomes worse when you realize that there are literally hundreds, if not thousands of visits to the United States over the course of an Administration. Interagency officials responsible for a priority administration program or working on headline-grabbing material are likely to be sequestered by these requests, diverting attention away from the execution of their programs. During our time in government, we would frequently find ourselves fending off such requests from other White House components or the State Department in the interest of protecting the time and resources of agencies in our respective domains. The urgency to provide something usually results in low-effort or empty commitments that are easily placed in contrast with those of foreign governments like the European Union or People’s Republic of China.
While we certainly understand the demand that administrations be able to produce examples of how they are delivering on the President’s priorities, it is important that such requests be properly staffed and resourced. When there are obstacles, the “center of government” organizations should provide sufficient support from champions to overcome those obstacles in order to ensure their successful completion. A good press release is seldom a substitute for the delivery of an actual result.
Conclusion
The persistent failure of interagency coordination is a systemic result of an administrative state that structurally prioritizes parochial autonomy of specific agency equities over collective efficacy. As demonstrated by the fragmented governance of space activities and the “zero-cost” compromises of underfunded committees, the current coordination framework incentivizes performative alignment rather than meaningful reform. These failures place an undue administrative burden on the public and stifle innovation and good governance.
To bridge the gap between policy aspiration and implementation, the federal government must push back against tribalism that currently defines interagency coordination. By barring advisory committees from delegating their responsibilities to the interagency, mandating rigorous economic transparency for new proposals, and ensuring that Congress provides the specific resources required for coordination, the Executive Branch can finally coordinate policy priorities. We’ve proposed that the federal interagency deliberative process requires a fundamental realignment of incentives, ensuring that the Government speaks with “one voice,” expressed through decisive action rather than just diluted reports, unfunded and unimplementable mandates, and perennially delayed policymaking. Such reforms will provide solid ground on which policy recommendations can be more safely landed.
A Plan for Revitalizing the U.S. Auto Industry
It’s January 20, 2029. The new President, committed to revitalizing America’s once-dominant car and truck industry, requests a briefing on the current state of affairs. The news isn’t great. Global market share of the big American automakers has bottomed out at single digits, well below the nearly 25% it enjoyed in the 1990s. Factories that made cars for export are either closed or closing.
That’s because the rest of the world doesn’t really want American cars any more. Continued tension and uncertainty in the Middle East has kept gas prices high and underscored the security and economic vulnerabilities of overreliance on oil. Outside the United States, consumers are snapping up Chinese-made EVs that go hundreds of miles, charge in minutes, and sell for less than $10,000. Industry is similarly pivoting to electric freight. On the home front, Americans are angry that U.S. policy is preventing them from buying exciting cars hitting the streets in neighboring Canada and Mexico. Workers aren’t much happier, as trade disputes and tariff barriers cause the once-integrated North American auto industry to come apart at the seams. And the U.S. truck oligopoly still runs on diesel, adding cost to almost every consumer good.
The President sighs and asks their team for a plan. Fortunately, they have one.
The World is Jumping Ahead on Electric Vehicles, Whether the U.S. Likes It Or Not
The writing is on the wall: electric vehicles (EVs) are the future. Industry knows it. Workers know it. Investors know it. Politicians on the left and right know it. EVs are simply getting too good, too quickly, for even the most pro-fossil policy agenda to stop. That’s good news for our climate, and for consumers globally, who deserve access to the latest, greatest, and cheapest cars. But it’s bad news for Americans, who are going to suffer the long-term effects of short-sighted transportation policies that fail to acknowledge economic and technological reality.
Eventually, this reality is going to become impossible to ignore. EVs are already dominating in countries as diverse as China, Norway, Vietnam, and India, and the geographic spread is only accelerating. Sooner or later, the United States is going to need a plan to catch up. It’s clear that what the plan won’t be is a simple return to the outdated regulatory frameworks we used for decades to regulate vehicle emissions and fuel economy. We need to think more broadly and more creatively.
Why Nixon-Era Statutes Won’t Catch Us Up On Their Own
Two statutes have historically governed vehicles. The 1970 Clean Air Act (CAA) structures the tailpipe emissions standards that the EPA and California enforce, while the 1975 Energy Policy and Conservation Act (EPCA), which charges DOT with setting separate (albeit overlapping) fuel economy standards. These laws, as we’ve previously written, were helpful for making gas-powered cars cleaner and more efficient, but they weren’t designed to guide a wholesale transition from fossil to electric technology with the pace and comprehensive scope needed.
There’s a case to be made for doing your best with old tools if those are the tools available. Indeed, that’s why the Biden administration’s vehicle regulatory regime was largely predicated on the CAA and EPCA. But these tools have become far less available in light of the second Trump administration’s actions. The Trump administration, in partnership with Congress, has dismantled the legal basis for regulating greenhouse gas emissions from vehicles under the CAA, gutted the fuel economy program that EPCA sets up, and rolled back California’s EV rules (which traditionally set the most stringent, future-forward vehicle standards). It also fired lots of people from federal agencies who understood how federal vehicle regulations worked. Thus from both a legal and a capacity angle, CAA and EPCA have become significantly weaker policy levers. Compounding these issues is the fact that the Supreme Court (now and likely into the near future) is profoundly hostile to regulatory ambition, limiting the probable reach of new rules set under these statutes.
Even if one could magically snap Biden-era vehicle rules back in place, those rules would only cover one player in the auto market – automakers themselves. Telling automakers that they have to develop cleaner, more innovative vehicles is an important tactic, but it is not a comprehensive policy agenda. CAA and EPCA don’t cover worker pensions, factory refits, trade policy, or continental industrial strategy. They don’t deploy charging networks or build out the grid. They don’t help fossil-fuel-producing counties diversify their economies, don’t structure smart spending priorities for the transportation system, don’t thoughtfully manage refinery closures and other giant infrastructure shifts. And as we’ve seen, CAA and EPCA don’t put real pressure on giant incumbent companies to change, and to make affordable new products, rather than to lobby hard against rules they don’t like.
By 2029, many of these core economic and industrial policy areas will also be in shambles. And as we’ve learned from the partial repeal of the Inflation Reduction Act, a party-line reconciliation bill won’t be a silver-bullet fix. Reconciliation bills are, as the Trump administration has made clear, no basis for stable investments. Fiscal policy is useful. Unstable budget policy will not draw in the billions in investment we’ll need to catch America back up.
These aren’t ancillary problems. Failing to manage the EV transition as the giant economic shift it actually is makes the policy and politics of the transition brittle and subject to reversal.
And Neither Will Ad Hoc Dealmaking
We need approaches that both compel and encourage giant incumbent companies to change and make affordable new products, rather than lobby hard against rules they don’t like. We need approaches that take the lived experiences of people who can’t afford to finance a new car, who don’t have access to charging at home, or whose livelihoods depend on the fossil economy seriously.
Yet the United States has never had a clear, cohesive, and comprehensive electrification and innovation strategy for the auto sector. In the absence of a clear national program, we’ve cut a series of tenuous deals to structure nearly twenty years of U.S. vehicle policy.
President Obama, during the 2009 auto bailout, was able to use fiscal recovery funds to steer the auto industry into accepting a first round of emissions rules from the feds and California. This “car deal” set the template for several more. The first Trump administration (with tacit support from automakers looking to slow EV investment) blew it up. California stepped into the breach with another set of deals, persuading companies to keep making EVs in exchange for regulatory relief and by threatening to stop buying government cars from holdouts. Team Biden then used Inflation Reduction Act funds to provide EV rebates and support EV manufacturing, as a spoonful of sugar to help a new round of federal and California emissions rules go down. And then the second Trump administration blew up those deals, again with industry support as American companies pivoted back toward short-term profitable gas SUVs.
Same story with trucks. California made a deal with the truck makers to continue progress on clean trucks. Post-Trump 2, those same truckmakers sued California to blow up their own deal. They are now backing Trump on his effort to destroy the entire CAA greenhouse gas vehicle program.
These deals were crucial progress at the time – and it would have been rational for the industry to keep its word. But it didn’t – policymakers shouldn’t be fooled again, or lulled into thinking that even good deals are a long-term substitute for durable policy. Relying on ad hoc dealmaking is an absolutely wild way to run a major industrial sector.
No wonder Chinese companies, able to rely on consistent government policy, have surged ahead while American consumers struggle to navigate the increasingly byzantine and expensive world that is the American auto market. While American workers move their families to be near good-paying auto jobs only to see investment pulled back and factories shutter. And while auto companies, in the absence of a stable and dependable policy environment, almost inevitably engage in short-term profit chasing rather than longer-term innovation.
A Path Back to the Future for U.S. Auto Policy
What breaks this bad pattern? Green industrial policy, embedded in a bipartisan statute that can pass a filibuster-proof majority of the Senate, and that blends competition and innovation.
Because we need both. The U.S. auto industry is highly concentrated, with only a few companies dominating domestic sales of both cars and trucks. These companies are used to calling the shots and setting prices, which is terrible for everyone but CEOs. American auto CEOs are deathly afraid of real competition – witness how Tesla’s new affordable electric semi is sending major ripples through the entire freight sector. But it’s been decades since the United States seriously enforced antitrust laws. Rather, politicians from both parties have raced to erect steep tariffs shielding the American auto sector from essentially all real global competition. There’s a role for any government to play in supporting its domestic industries. But that role isn’t propping up failing legacy companies indefinitely, at the public’s expense.
A well-designed statute could strengthen market oversight and competition while also providing American companies with reasonable support. Don’t like the Chinese-backed EVs that are undercutting your market? Start making and selling better, cleaner, and cheaper vehicles – and we’ll help. This is the logic that motivated Canadian Prime Minister Mark Carney and Mexican President Claudia Sheinbaum to welcome Chinese competition while bolstering their domestic industries. The U.S. can catch up with the same approach. For instance, government can readily provide loan funds and policy support for overseas companies that want to scale up in the United States, in partnership with domestic manufacturers. Such joint ventures are win-wins: the foreign company gets a new market and learns how to build for the U.S. market; the domestic company gains process knowledge and the IP it needs to modernize its products. That is exactly what Toyota and GM did a few decades ago, when the U.S. industry faced major competition from overseas autos (then, from Japan). The strategy can work again, for cars and trucks and batteries. Throw in better federal loan terms for union facilities, and you have a worker-friendly, industry-friendly, pro-competition, pro-consumer package. And this time the government can even explore taking an equity stake. Because why not re-shore profits, too?
Is comprehensive statutory reform ambitious? Certainly. But anything less consigns core national economic and industrial strategy to the vagaries of judicial interpretations of decades-old pollution statutes. Half-measures will, given the context above, be essentially useless. As with riding a bike, we can’t afford to pedal slow. The fate of a vast national industry warrants real national vision, rooted in a democratic vision for ordinary people. Democracy repair is climate repair is industrial reconstruction.
To deliver that vision when the policy window emerges – when the next President asks for it – we’ve got to start structuring it now. We suspect it will include some or all of the following elements:
- A national electrification trajectory, established in law. No more trying to retrofit EV rules from fossil-era statutes. We need a clear, negotiated path for electrifying America’s auto sector and restoring our global competitiveness, with related provisions on battery life and durability. This path must be binding on manufacturers.
- An accelerated charging network. There’s truth to the industry argument that they can’t sell EVs without chargers. We need federal funding to fill gaps in the charging network that private capital won’t (taking inspiration and precedent for federal funding for rural electrification), while incentivizing additional private-sector buildout guided by federal reliability standards and provisions for incorporating chargers in building codes and urban planning requirements.
- A continental industrial strategy. North American EV manufacturing should be baked into the USMCA and trade policy. Do we focus on advanced manufacturing of batteries? On assembly? On new high-tech models? We don’t have to do it all, but we do have to decide and put real resources and legal muscle behind it.
- Trade policy that lowers prices and creates new manufacturing. Our current system both blocks affordable Chinese EVs with prohibitive tariffs and fails to transform domestic industry. Explore options like joint ventures that on-shore competition through domestic-foreign partnerships, paired with tariff adjustments tied to domestic policy supports.
- A real free market for vehicles. Today’s American vehicle market isn’t free. The few companies that dominate vehicle sales in America serve consumers poorly yet get bailed out when they fail to compete. Enough. It’s time to provide Americans with real choices. That means, at minimum, statutory terms ensuring that prices are transparent and fair (as California is currently considering) and resources for anti-trust enforcement and consumer protection.
- Genuine support for workers. Federal support (including funding, loan guarantees, land use and permitting policy, and offtake opportunities like fleet procurement) should be conditioned on high-road job standards (e.g., labor neutrality agreements, training programs (including apprenticeships and new career opportunities), community benefit agreements, and pension protections. This policy should move in tandem with pro-worker reforms to labor law, such as the PRO Act.
- Genuine support for communities. Local governments and communities seeking to attract new manufacturing, manage plant closures and repurposing, or diversify revenue streams will need support. Federal government can provide funds to patch short-term budget holes as well as technical assistance and capacity-building grants to manage these issues across silos – all while deeply engaging state and local leaders to co-create and iterate national auto strategy.
- Innovative fiscal mechanisms. Public money should be used to align and leverage private capital into serious modernization of the U.S. auto sector. The federal government can provide loans for new auto facilities and market hubs, make loans for EVs (especially expensive medium- and heavy-duty vehicles) as standard and easy to secure as loans for other vehicles, push prices lower by tying federal incentives to reduced MSRPs, and even consider funding some of these measures by taking equity stakes in firms it funds. There is plenty of private capital in the United States to harness towards a globally competitive auto sector. The role of public funding is to push it in the right direction.
- Durable implementation support and education. We’ve seen how the IRA failed to build a political constituency because it rolled out too slowly, and with too little political cover. This time, the national program needs to come with durable support to increase implementation capacity at all levels, for speed, and a clear political plan to keep the American people engaged on this crucial national project, sharing its rationale and working through both concerns and victories as they come.
What Needs to Happen Now
The comprehensive, statutorily based plan we envision is only so much wishful thinking today. But we can actually create the conditions to realize this plan in the future. That’s because states have very substantial economic development authority, complemented by the substantial financial muscle of large state-based green banks and infrastructure banks.
What if the next governor of California provided state-backed loans for any company seeking to expand an EV or battery firm with pro-union policies in the state? The governor might particularly encourage joint ventures, perhaps looking to the Chinese firm BYD’s existing facility outside of Los Angeles (which doesn’t make cars and semis now, but could be expanded to do so). This isn’t wishful thinking. California more or less created Tesla with over $3.2 billion in subsidies. It can now create a host of new competitors, but this time it can also be taking equity to ensure profits flow back into a “transition fund” in the budget that supports economic growth in counties facing declining oil production. It can simultaneously pass state laws strengthening oversight of the U.S. auto industry, forcing transparent prices and pro-competition market structures.
If California leads, it’s unlikely to stand alone. States like Michigan might well jump in, using its existing Office of Future Mobility and Electrification to draw in billions in investment in existing facilities and pairing public investment with scaled-up private capital in powerful capital stacks. So might Texas, Illinois, Ohio, and Georgia – states that have made significant investments in EVs and batteries that they, and their constituents, are loath to lose. After a year or so, we might reasonably expect an MOU linking a growing state coalition that is working to develop the capital instruments and policies needed to grow industry.
With this policy certainty, investments start flowing at scale and a positive cycle begins. States continue to implement the panoply of policies in their control that otherwise support economic modernization, from clean air planning to freight system electrification rules and programs to policies that help vehicle batteries profitably support the grid by storing energy. They update building codes, deploy chargers, insist on durable and quality products using their consumer protection laws, and steer public money towards necessary infrastructure. Critically, they root all that work in expanded capacity to navigate the “messy middle” of the ongoing clean-technology transition, ensuring that neither workers nor communities will be left in the lurch.
At the same time, far-sighted public officials, backed up by civil society and foundation support, start bringing together the interests the new President will need for a major statutory play. They bring in counterparts from Canada and Mexico, recognizing that the continent has to either work together or lag together. Soon, the collaboration starts looking like an actual industrial strategy at the scale of East Asia’s behemoths.
In this scenario, when the next President calls for an auto modernization plan, Governors across the country back her. So do many major investors, donors, local officials, and federal electeds. After all, they’ve already seen the future in their states, and it is in their interest to scale up the national program. When the gavel falls in the first year of the 121st Congress, the conditions for change are in place, and twenty years of shaky rules and ad hoc deals give way to a new approach – a lasting one.
Four Innovations Driving Climate Progress in State Government
Subnational governments—cities, states, and counties—took some of the earliest steps in the United States to protect air quality, water quality, and public health. Over the last several decades, as the federal government has wavered in its commitment or failed to take a leadership role on climate change, states and cities have continued to act, both by bold pronouncements and aggressive targets and in more quiet, subtle ways that have eased the path for clean energy and other investments. This has resulted in a diverse cohort of cities and states that have made great progress in advancing clean energy and other climate solutions and are well-positioned to continue this leadership and innovation.
However, the federal government is taking aim at subnational authority and leadership, including threats to states’ actions deemed “overreach.” This threat from the federal government not only limits the ability of subnational governments to develop innovative tools to address climate challenges, but can hinder opportunities for economic development by limiting access to lower cost energy solutions, stifling innovation in emerging industries, and hampering global competitiveness.
Subnational Governments as Innovators and Leaders
Subnational governments, in liberal and conservative states, have long been the drivers of clean energy and climate progress in the United States.
Iowa adopted the first renewable portfolio standard in the United States in 1983, which laid the foundation for the state to remain one of the top renewable energy producing states in the country. George W. Bush and Christine Todd Whitman took aggressive actions as governors to reduce greenhouse gas emissions. Like Iowa, this early commitment in Texas has proven durable as the state continues to lead on renewable energy and energy storage deployment – due in large part to the state’s work to accelerate deployment of renewable energy projects, including permit streamlining and speeding interconnection for new projects. Renewable electricity generation reached a new high in the United States in 2024 and four of the top five producing states were “red” states .
U.S. cities and states have also taken a lead to affirm global commitments to achieve greenhouse gas emission reductions. In 2005, the U.S. Conference of Mayors launched its climate commitment when 141 mayors committed to meeting the emission targets included in the Kyoto Protocol. That same year, Governor Arnold Schwarzenegger issued an executive order in California, establishing economy-wide greenhouse gas emission reduction targets for 2010, 2020, and 2050.
Subnational actions are far more than symbolic. As of today, 33 states have climate action plans, 24 states have economy-wide greenhouse gas emission reduction targets, and 36 states have renewable energy or clean electricity standards. These commitments spotlight state leadership and serve as a reminder of the role of states’ authority and commitment to environmental leadership and their ability to serve as a backstop to federal inaction. They are important in several important ways. These commitments to reduced emissions and clean energy targets send a signal to developers, investors, and other governments that they have customers, markets, and willing partners. This has driven investment in companies and created opportunities for workers and industries in these states.
Subnational leadership is equally important for reimagining the systems, governance, and institutions to make these targets, goals, and investments a reality. For example, cities and states have developed streamlined permitting and inspecting processes for residential solar and energy storage installations (167 jurisdictions in 47 states), identified least conflict areas for large scale renewable energy projects (California and Washington), and developed innovative finance structures and institutions to support clean energy investment and development (e.g., Green Banks in 29 states, the District of Columbia, and Puerto Rico). Scaling these innovations can accelerate the energy transition and boost economic development, workforce development, and local communities.
The Need for Subnational Leadership
Subnational leadership on decarbonization is a practical necessity. Modeling shows that meeting global emission reduction commitments requires actions by subnational governments and businesses. In the United States, existing commitments by subnational governments and businesses could reduce national emissions 25% below 2005 levels by 2030. State level action can be approximately cost-comparable to federal (top-down) action and it tends to focus more on electrification of energy end uses, clean energy, and direct air capture, all solutions that fall under the authority of subnational.
Subnational governments hold primary authority over infrastructure development, siting and development of energy generation and transmission, energy efficiency in buildings, and land use decisions that determine development and conservation patterns. For example, scaling electrification to levels needed to achieve climate goals requires massive build out of clean energy resources and installation of heat pumps and other electric appliances at the household level. At the same time, state and local economies and communities are intimately connected to legacy industries that have shaped local fiscal structures, workforce, and cultures. Therefore, realizing the transformation necessary to decarbonize requires strategic action by state and local government.
Current regulatory structures are not optimized to navigate the challenges of decarbonization. Decarbonization is a systems challenge that depends on successful transformation of technical, social, and economic systems. Decarbonizing the energy system requires building new, clean energy and transportation systems, while at the same time making the investments needed for an orderly transition away from carbon in legacy industries like oil and gas. This dual approach enables environmental progress, while also protecting the workers and communities reliant on legacy industry. This requires linking environmental goals and policy in alignment with economic development, industrial policy, and environmental justice and equity goals. Failing to take an integrated approach repeating patterns of earlier transitions that concentrated pollution and contributed to growing inequity and environmental justice problems. Subnational governments are well positioned to take this integrated approach.
Working at a state or local level means that policies can be tailored to meet local contexts (e.g., economic, political, or social) in a way that top-down federal policies cannot. This is especially important because “successful implementation” means more than reducing emissions. Successful implementation means meeting climate and environmental goals while promoting prosperity and equitable opportunities for all residents, businesses, and communities. Success depends on accelerating project implementation while also addressing affordability issues and promoting society-wide benefits. Successful implementation stories are needed across a diversity of places to demonstrate approaches that are easy for others to follow and will resonate with cities and states that face different political, economic, and social situations. State and local government provide the right scale for building these solutions.
Challenges to Subnational Leadership and Innovation
It must be noted, especially at this point in time, that while the United States has a strong tradition of subnational leadership, it is not guaranteed. Subnational governments are experiencing many challenges that threaten to erode their leadership position—some inherent in the nature of the energy transition and other external factors.
A challenge of the energy transition is the need to build up new, clean energy systems while also carefully phasing out old, polluting systems. Governments need to accelerate investment in new clean energy, while continuing to invest in legacy industries. Failing to do both can introduce threats that can erode or even stall progress on decarbonization. California is experiencing this challenge now as the state navigates the impact of reduced demand for gasoline, a success of the state’s clean transportation policies, and what that means for the state’s oil and gas industry. The instability for oil and gas has resulted in the planned closure of two of the state’s refineries. While these closures will reduce pollution in host communities, they will also result in lost jobs and revenue and have the potential to increase the price of gasoline for California consumers. These dynamics have required the state to take actions to stabilize the oil and gas industry, while also accelerating its clean energy investments.
These challenges, inherent to the transition, are being exacerbated by other factors that threaten to erode subnational leadership and innovation, including:
- Rapid policy shifts at the federal level including termination of federal funding for clean energy projects, permitting uncertainty, and other disruptions that create significant uncertainty for governments, project developers, and labor.
- Growing inequity resulting from systematic racism, economic structures, and lack of opportunity poses a risk that some communities and populations will be left behind in the energy transition.
- Attacks on subnational authority to establish and implement climate, energy, and environmental policies.
While daunting, these challenges make the need for subnational innovation more important than ever.
Current Opportunities for Subnational Leadership and Innovation
Now, more than ever, subnational governments need to be places for regulatory ingenuity and action in the face of strong headwinds. The urgency of climate change requires immediate acceleration of the implementation of climate solutions—to reduce greenhouse gas emissions, protect public health and wellbeing, align economic development with environmental goals, and build resilience to changing climate and extreme events. Cities and states are best positioned to design policies to accelerate clean energy, innovation, and economic development because they can design approaches that work in different social, political, and economic contexts.
Innovation lies in the “how”—how to scope the challenges and design solutions that recognize the complexity of the decarbonization challenge. Subnational governments have already demonstrated several the power of regulatory innovation in several areas that show how rethinking regulatory and governance systems can accelerate progress:
- Innovation 1: Least conflict siting
- Innovation 2: Automated permitting
- Innovation 3: Making projects work for all
- Innovation 4: Deploying innovative approaches to funding and finance
These innovations by subnational governments show how government can accelerate the deployment of clean energy and other climate projects, implement projects that work in specific contexts, and deliver benefits to people and local economies.
Innovation 1. Least Conflict Siting
Uncertainty, conflict, and complex permitting and siting processes are major impediments to project implementation. Developing new approaches to siting and permitting can reduce uncertainty and delays that can increase project costs and diminish developer confidence. State and local governments can develop and deploy several innovative tools that can make it easier and less expensive to implement climate solutions, while also increasing transparency and engagement.
Cities, states, and counties can improve the siting and permitting processes by removing barriers to implementation, engaging with diverse stakeholders, and reducing costs for developers, government, and residents and businesses. For siting, this can include thinking at a regional or multi-project scale to identify priority areas for development. Engaging stakeholders early in the process can reduce objections and challenges later in the project process. Least-conflict siting processes provide one approach to innovate in the siting process.
Siting Innovation: Least Conflict Siting Process
Least conflict siting is a data-driven, participatory siting process guided by stakeholder priorities. A least conflict siting process uses spatial data that reflect stakeholder priorities (e.g., prime agricultural lands, sensitive habitat, etc.) to identify areas for infrastructure siting that avoid areas of high conflict. Using a participatory, stakeholder-driven process can avoid conflict at later stages in the development process, reduce uncertainty for developers, and provide a more transparent process for local residents, business, and other stakeholders. By identifying least conflict lands, stakeholders can then focus on removing obstacles to development on those lands (e.g., access to transmission). The process is non-binding, so can be adjusted as conditions or priorities change.
A least conflict siting process has been used in two regional contexts. The Center for Law, Energy, and the Environment and the Conservation Biology Institute piloted a least conflict siting approach for solar energy development in the western San Joaquin Valley in California in 2016. The project aimed to identify areas with least conflict for renewable energy development in a six-month process. A least conflict siting process has also been piloted for the Columbia Plateau in Washington. The project spanned eight months and concluded in 2023.
Both processes used geospatial data made accessible to all stakeholders through a collaborative gateway. Following this pilot program, Washington State passed a law in 2023 to improve project siting and permitting includes this least-conflict approach as a tool to be referenced for large scale renewable projects.
While this approach was developed in the context of large-scale renewable development, a least conflict-type process can be applied to a range of project types. This could include minerals mining, carbon removal, or transmission projects. The least conflict approach surfaces priorities and concerns in each region and the results of the process can be applied to different types of climate and energy projects, providing an opportunity to provide efficiency. A least conflict siting process requires commitment from a local permitting authority (e.g., local government), project developers, and stakeholders, including environmental, business, economic development, and other groups. The process also requires some investment to establish a robust process, including: spatial data showing energy, environmental, and other characteristics on the landscape, robust engagement, and strong facilitation.
Innovation 2. Automated Permitting
Delays in permitting increase project costs for developers and households. Developing transparent and simpler approaches to permitting can reduce delays, errors, and costs. Analysis of rooftop solar permitting in the United States shows that nearly 80% of a system’s cost is attributable to “soft costs.” These include design, project management, permitting, inspections, and interconnection. Reducing these soft costs through streamlined and/or automated processes can significantly reduce the costs of these projects. Streamlining can address many stages of the permitting process, including application, evaluation, and inspection – saving time and money for applicants, installers, and permitting agencies. For large-scale projects, mapping the permitting process to identify opportunities for creativity, flexibility, and efficiency can improve the permitting process.
Permitting Innovation: Automated Permitting and Inspection
Cities and counties are the permitting authorities for many clean energy projects, including rooftop solar and storage systems. Permit Power is a U.S.-based non-profit organization focused on reducing the bureaucratic impediments to deploying residential solar and battery storage. The organization’s research finds that a typical residential solar installation in the United States is up to seven times more expensive than a similar installation in Australia or Germany. To address this disparity, Permit Power is working at the state and local level to advance permitting and interconnection reform to reduce barriers to residential solar and battery storage projects.
Developers have identified permitting and inspection as major barriers to residential solar and storage project deployment—increasing both the cost and timeline for projects. Several states have adopted laws allowing or requiring cities and counties implement automated or streamlined permitting processes, while others have opted in on their own. The state laws that encourage or require automated permitting for solar projects have taken different approaches, generally with a nod to maintaining flexibility. New Jersey’s law directed a state agency to develop an automated permitting platform, but also to allow local jurisdictions to adopt an alternate platform with oversight from the state agency. Texas and Florida passed laws allowing the use of automated platforms but not requiring them. Maryland passed a law requiring local jurisdictions to use an automated permitting platform.
The Solar Automated Permit Processing Plus (SolarAPP+) provides a free, widely applicable platform to streamline permitting for rooftop solar and solar plus storage projects. SolarAPP+ is available free to jurisdictions designed to make the installation process easier for contractors and permitters, reducing needed staff resources, project timelines, and permitting delays. The National Renewable Energy Laboratory (NREL) developed SolarAPP+ in collaboration with industry and building inspectors. The platform streamlines permit approval for installations that meet specific requirements.
SolarAPP+ was launched in 2021. At the end of 2023, 167 permitting jurisdictions have adopted or piloted use of the SolarAPP+, and close to 600 additional jurisdictions have expressed interest in the application. Annual evaluations of the platform’s use show that permits for code-compliant systems is nearly instantaneous and that SolarAPP+ projects complete the full permitting process faster than installations that use the traditional permitting process. The evaluations also document savings in staff time and reductions in project delays.
SolarAPP+ is now managed by an independent foundation and is available to all jurisdictions free of charge.
Innovation 3. Making Projects Work for All By Using Community Benefit Tools
A risk of moving projects at a more rapid pace is the potential for harmful, unintended impacts on communities and the environment, including concentration of industrial activities, damage to habitat and natural systems, and reductions local quality of life. At the same time, these projects can bring economic development, workforce, and associated benefits to host communities. Community benefits tools can reduce conflict and improve project delivery by minimizing harms and harnessing benefits from these investments.
Community benefit tools can provide a mechanism for ongoing accountability and transparency for host communities, businesses, residents, and other stakeholder groups. These tools include community benefits agreements, community and cooperative ownership structures, and community oversight structures. If carefully crafted, community benefits tools have the potential to deliver meaningful benefits to infrastructure host communities, provide opportunities for community oversight and shared governance of projects, and reduce friction between developers and communities. Including community organizations and stakeholders as planning and implementation partners is vital to the success of these tools.
Subnational governments can require or create incentives for the development and use of community benefits tools for project deployment and they also have an important role to play in building community capacity to engage in the development and deployment of community benefits tools. They are also well-positioned to create the guidance and accountability tools to create the conditions for more effective community benefits structures. However, the existence of a community benefits agreement or related tool is not sufficient in and of itself, these tools need to be developed and designed well to deliver benefits to communities.
Importantly, linking community benefits to siting and permitting innovations can provide durable assurances of project performance and that a project will deliver benefits to a host community.
While requirements and incentive structures have promise, it is critical that tools are available to ensure that community benefits agreements are done well. These include guidelines for agreement development and technical and legal assistance for communities to ensure that agreements deliver real benefits to communities. It is worth noting that developing project by project community benefits agreements could result in two unintended and undesirable outcomes: the added process could slow down or discourage wanted projects and host communities end up with piecemeal benefits that cannot deliver meaningful and transformative investments in a community (e.g., comprehensive workforce development, integrated infrastructure investments, etc). Imagining scaled approaches (e.g., across a city or county scale) to deliver community benefits in a holistic manner across multiple projects in that area could increase efficiency for developers and support transformative investments in places hosting multiple projects or project elements.
Community Innovation: Models to Deliver Community Benefits
Community benefits agreements associated with development projects can deliver meaningful benefits to host communities, including investments in infrastructure, workforce development, and other community investments. State or local governments can require community benefits frameworks for projects in a specific geography (e.g., a community benefit ordinance) or create incentives for the development community benefits agreements or other structures to streamline project development (e.g., California Assembly Bill 205).
Detroit adopted a Community Benefit Ordinance in 2016. The ordinance requires that projects valued above $75 million or that receive significant subsidies from the city provide additional benefits to the community where a project is sited. When the ordinance is triggered, a Neighborhood Advisory Council from the project’s impact area is formed to work directly with the developer. The City of Detroit tracks progress on the commitments made through the agreements developed under the ordinance. Since its passage, eleven projects have finalized agreements under the ordinance and four more are in progress. Regular review of the ordinance’s performance has identified areas for improvement but monitoring shows that it has delivered measurable benefits in Detroit communities.
In accordance with California Assembly Bill 205 (2022), the California Energy Commission (CEC) has developed an Opt-In Certification program for clean energy projects including large-scale renewable energy (i.e., greater than 50MW), energy storage, and some clean technology industrial facilities. Through the opt-in certification program, the CEC can issue a permit for the project and enable it to forego permitting by local land use authorities and most, but not all, state permits. To qualify for the Opt-In Program, a project must meet a set of requirements, including entering into at least one legally-binding and enforceable agreement that benefits one of more community-based organizations in the project area (e.g., a community benefits agreement). For most qualifying projects, the Opt-in Program provides a faster timeline for environmental review (within 270 days of a project’s complete application, in most cases). However, implementation of the Opt-In Program is limited to date.
Innovation 4. Develop Innovative Financing Tools
Funding and finance for climate actions is a major barrier to advancing action, especially as the federal government claws back and reduces federal funding for energy, environmental, emergency response, and other programs that states, cities, and project developers have depended on. Now more than ever, developers, cities, and states need to be innovative in how they access and deploy funding and finance tools to support project development.
Subnational jurisdictions can initiate various revenue generation strategies to support project development. They can also access or establish different funding (i.e., grants) and financing strategies (i.e., loans) to support public and private project development. Subnational revenue generation tools include bonding authority, taxing structures, credits programs, and implementation of pricing programs (e.g., congestion pricing in New York City). They can also establish and/or access financing institutions like green banks and public-private structures to finance project development.
Subnational governments need to deploy a suite of revenue generation, funding, and financing strategies to support implementation and unlock access to private capital and investment. This is especially true given current threats to municipal finance, including withdrawal of federal funds, increasing climate risks and disasters, and the fiscal dimensions of the energy transition that affect local revenue structures. An analysis of options to support implementation of San Francisco’s Climate Action Plan found that the City needed to access all tools available to it to achieve the levels of investment necessary to implement the plan. This included development of new tax and fee structures, use of financing districts, integrating climate actions in the City’s schedule of general obligation bonds, establishment of a green bank, and implementation of pricing policies, including congestion pricing. Since the time of the analysis, the City of San Francisco passed Proposition A in March 2024, the city integrated climate-related actions into a scheduled general obligation bond to support affordable housing.
Financing Innovation: Public Finance for Transmission Infrastructure
Thirty states either have or are considering development of a green bank. A green bank provides access to capital for clean energy and other sustainable projects by issuing loans to projects that might otherwise have difficulty accessing capital. Programs within the California’s Infrastructure and Economic Development Bank (I-Bank), the State’s financing institution to support public infrastructure and private development projects that benefit California’s economy and quality of life, serve as the state’s green bank. Recent legislation established a program within the I-Bank to support investment in transmission infrastructure.
Transmission infrastructure is needed in many regions to distribute clean, renewable energy from where it is generated to load centers. California anticipates a quadrupling of in-state renewable energy generation by 2045, which will include offshore wind generation off the northern and central coasts, large scale solar generation in the Central Valley, and geothermal energy from the inland south regions of the State. Currently new transmission is funded though investor-owned utilities that pass costs on to ratepayers or by private developers.
To provide an alternative model, California recently established a public financing mechanism for new transmission infrastructure, the California Transmission Accelerator Revolving Loan Fund Program. The Accelerator will exist in the State’s I-Bank, the State’s financing institution to support public infrastructure and private development projects that will benefit California’s economy, jobs, and quality of life. The Governor’s Office of Business and Economic Development will develop a financing and development strategy in coordination with the State’s energy agencies to guide the implementation of the Transmission Accelerator. The program is designed to reduce burdens on ratepayers by using State funds to support needed transmission infrastructure development.
Closing Thoughts
We are at a critical moment for climate progress—given both the urgency of climate change and political polarization in the United States. However, this combination provides an opportunity for creative thinking and innovation at the subnational level. State and local governments have an opportunity, and perhaps obligation, to reimagine the regulatory, institutional, and governance structures to design decarbonization strategies that work for state and local economies, communities, and the environment. If undertaken at scale and implemented quickly, these subnational actions can have a significant impact on carbon emission reductions.
Some ways to help realize this innovation include:
- Build Subnational Capacity. Cities and states need additional resources and capacity to develop policies and programs, access funding and financing to implement projects, and partner with industry, communities, and labor to build durable solutions—especially in rural and under-resources areas. Building additional capacity requires creative use of existing public resources; partnership with the private sector, non-governmental organizations, research institutions, and philanthropy; and building networks for peer to peer learning, sharing, and collaboration.
- Leverage Networks to Replicate and Scale Successes. Subnational governments have strong platforms to share and scale successful policy actions. When President Trump withdrew the United States from the Paris Agreement in 2017, a bipartisan group of states established the U.S. Climate Alliance. The members of the U.S. Climate Alliance have committed, collectively, to achieve net-zero emissions as soon as possible and by 2050 at the latest. Climate Mayors, established in 2014, is a network of almost 350 mayors from nearly all fifty states that are committed to upholding the Paris Agreement. Together, these networks—and others—represent a significant share of the U.S. population and economy and provide a community of practice to support policy development and implementation. Subnational collaboration extends beyond the United States, with subnational networks like the Under2 Coalition that includes over 180 subnational governments across six continents committed to achieving deep GHG emission reductions. These networks can provide durable platforms for replication and scaling of successful policy innovations.
- Respect the Power and Potential of Subnational Governments. Subnational governments have a long legacy of leadership and innovation, due in large part to the country’s history of cooperative federalism. This system has provided room for creative solutions and policies tailored to each state’s unique economy, environment, and culture. Congress and the White House need to respect this and build capacity in cities and states to enable them to align economic and environmental goals.
Outcome-Based Contracting Reorients Government IT Acquisition Around Public Value and Mission Results
The effectiveness of federal programs is increasingly determined by the technology that powers them. Yet decades of oversight and research have documented persistent challenges in large-scale IT modernization. The Government Accountability Office has repeatedly designated federal IT management as high risk, citing cost overruns, schedule delays, weak requirements management, and inadequate oversight. Bent Flyvbjerg’s research shows that large public-sector technology and infrastructure programs are especially prone to failure due to scope creep and cumulative risk. The Defense Innovation Board similarly concluded in Software Is Never Done that long development cycles and early requirement lock-in expose missions to unacceptable risk.
Across these analyses, the pattern is consistent: requirements are defined too early and too rigidly; performance is measured too late; incentives reward milestone completion rather than operational outcomes; and risk accumulates until deployment. These failures reflect several structural challenges—fragmented funding, leadership turnover, legacy system complexity, and acquisition models that delay validation and limit adaptation.
Traditional acquisition approaches assume stable requirements and predictable environments. Software-intensive systems do not behave this way. Requirements evolve, dependencies emerge during implementation, and technology ecosystems shift over the life of the contract. In this context, specification-driven models can increase risk by delaying feedback and limiting course correction.
This paper examines Outcome-Based Contracting (OBC) as a model for aligning acquisition with the realities of modern IT delivery. OBC reframes procurement around the staged achievement of measurable mission outcomes rather than the delivery of predefined technical artifacts. OBC ties funding, evaluation, and continuation decisions to mission outcomes and pairs naturally with iterative delivery practices that surface and reduce risk early.
Outcome-Based Contracting
Federal acquisition models have evolved over time in response to changing technologies and risks. Early approaches emphasized detailed specification and cost control, with contracts structured around defined requirements and reimbursement of inputs (e.g., cost-plus and fixed-price models). As systems grew more complex, performance-based contracting emerged to shift focus from activities to measurable outputs and service levels. However, in complex and dynamic environments, even performance-based models often remain tied to predefined deliverables and intermediate metrics, limiting their ability to adapt as conditions, requirements, and understanding evolve over time.
Outcome-based contracting (OBC) represents a further evolution. It structures the government–contractor relationship around shared accountability for mission results rather than delivery of predefined outputs. Its defining feature is not a pricing model, but the alignment of incentives, governance, and performance measurement around measurable mission outcomes.
As Allan Burman notes, building on performance-based contracting, OBC shifts accountability from activities and milestones to mission outcomes. In practice, it establishes a structured process in which government and contractor jointly deliver measurable results, with contracts defining decision rights, evaluation mechanisms, and adaptive processes.
Key features include:
- Shared accountability: success is defined in operational terms, not artifact delivery
- Collaborative outcome definition: the government defines the problem to be solved, contractors propose and refine approaches as evidence emerges
- Adaptive performance management: metrics guide decisions, not just compliance
- Joint problem solving: governance supports rapid adjustment when performance diverges
A useful way to understand outcome-based contracting is as a managed performance relationship rather than a one-time procurement transaction. As research from the IBM Center for The Business of Government emphasizes, effective outcome-based models require clearly defined desired results, measurable indicators of success, and ongoing performance management processes that allow both parties to assess progress and adjust course. This includes establishing baseline performance, continuously monitoring results, and linking financial incentives, contract options, and governance decisions to demonstrated improvement. Critically, these models depend on sustained collaboration and transparency: agencies must be able to interpret performance data and engage in joint problem-solving with vendors, rather than relying solely on compliance reviews. In this sense, OBC is not simply a different way to write requirements—it is a different way to manage delivery, in which measurement, incentives, and decision-making are continuously aligned to achieving mission outcomes.
Applying Outcome-Based Contracting to IT Modernization
Applying OBC to IT modernization requires three shifts: defining measurable outcomes, structuring decision rights, and organizing contracts around incremental delivery.
Defining outcomes
Mission objectives must be translated into measurable operational indicators—such as transaction completion rates, time to resolution, system availability, or error reduction. These indicators must be precise enough for evaluation while reflecting real-world service performance.
Effective models distinguish between:
- Mission-level outcomes (stable): e.g., reducing time to receive benefits
- Implementation metrics (adaptive): e.g., response times or interim system thresholds
For example, a call center contract might set a mission outcome of reducing resolution time by 30 percent, supported by metrics such as speed of answer, first-contact resolution, and callback completion time.
A central design question is how outcomes are embedded in the contract. Outcomes can function as binding accountability anchors, linked to evaluation, incentives, and option decisions, but not as rigid end-states. This approach is only effective when supported by governance structures that allow agencies to interpret performance and adjust delivery.
Critically, outcomes and the underlying problem definition must be treated as testable and subject to refinement. Initial problem framing is often incomplete in complex systems. Contracts and governance models should therefore include regular check-ins, using data, user research, and operational feedback to assess whether the problem is being solved as intended. Where necessary, agencies and vendors must be jointly empowered to restate or refine the problem to ensure continued alignment with mission needs.
Structuring decision rights
OBC requires clear decision making authority over priorities and tradeoffs. In software delivery, this centers on a strong government Product Owner (PO) role. The PO is responsible for backlog prioritization, acceptance criteria, and aligning delivery with mission outcomes. The PO must be empowered to continuously adjust priorities based on user needs and performance data without requiring contract modifications. Contractors are accountable for delivering measurable progress, but do not control mission priorities.
Governance must reflect agency maturity, and also the nature of the initiative. More mature organizations can rely on PO-driven execution and adaptive metrics, using contract outcomes as high-level anchors. Even in less mature agencies, OBC principles can be applied in targeted ways—particularly in user-facing systems or components where outcomes can be clearly measured. In some cases, especially large enterprise system implementations, hybrid approaches may be required. These may combine clearly defined objectives and outcome metrics with more structured implementation phases for core platform rollout. The key is not strict adherence to a single methodology, but aligning decision rights, outcomes, and delivery approach to the realities of the system being implemented.
Structuring incremental delivery
Contracts must support incremental, evidence-based delivery. Large, multi-year programs defer risk discovery until late in the lifecycle. Iterative delivery reduces this risk by shortening feedback loops: capabilities are deployed incrementally, evaluated under real conditions, and adjusted early. Incremental delivery provides disciplined mechanisms for iteratively paying down risk.
OBC complements this model by tying funding and continuation decisions to demonstrated performance. Agile practices surface risk; OBC aligns accountability and resources to its mitigation.
This has direct implications for funding models. Effective OBC implementations require upfront decisions about how much funding is allocated to a product or service, with mechanisms to adjust that funding over time based on performance. Budgeting should support iterative scaling—expanding or contracting investment based on whether outcomes are being achieved. This, in turn, requires financial flexibility, such as capability-based budgeting, and the ability to reallocate funds or leverage working capital-like mechanisms.
In practice, appropriations constraints can limit this flexibility. For example, agencies operating under single-year appropriations may struggle to dynamically adjust funding in response to performance signals. Addressing this requires coordination between acquisition, product, and financial management functions to ensure that funding structures align with the adaptive nature of outcome-based delivery.
Outcome-Based Contracting In Practice
Outcomes-oriented approaches are not new but remain underutilized in IT acquisition. Existing models demonstrate the value of aligning funding to measurable performance.
Within government, the Department of the Navy’s World Class Alignment Metrics (WAM) evaluates IT investments based on outcomes such as resilience, customer satisfaction, and cost per user. Similarly, Department of Defense Performance-Based Logistics ties compensation to readiness outcomes, and NASA’s Commercial Crew program links payments to demonstrated capability.
These examples share a core principle: funding follows validated performance rather than predefined inputs. Applied to IT modernization, this requires pairing mission outcomes with iterative delivery, clear decision rights, and sustained technical engagement. Without these elements, outcomes risk becoming abstract goals rather than operational tools.
Despite its advantages, outcome-based contracting is not the default in federal IT acquisition. In practice, existing incentives continue to favor specification-driven models: funding structures are rigid, oversight emphasizes compliance with predefined requirements, and procurement processes reward detailed up-front definition over adaptive execution. The following case illustrates how these dynamics shape real-world outcomes—and how leadership, governance, and delivery choices ultimately determine whether programs succeed or fail.
Case Study: SSA Call Center Modernization
The Social Security Administration (SSA) operates one of the largest public-facing service platforms in the federal government, serving approximately 70 million Americans through its national 800-number network and field offices, processing high volumes of calls. In 2017, the SSA faced growing problems with its aging, complex telephone infrastructure and rising wait times for the tens of millions of Americans who rely on the agency’s national 800-number for assistance with benefits, Social Security numbers, and other services. To address these issues, SSA launched the Next Generation Telephony Project (NGTP), a large IT modernization effort intended to replace legacy telephone systems and unify call handling across the agency.
NGTP emerged from a traditional acquisition model: a detailed, waterfall-style specification, a large systems-integrator contract, and milestone-based progress tied to predefined technical requirements. In February 2020 SSA awarded an IDIQ contract to Verizon to design, implement, test, transition, operate, and maintain the new telephony platform, including procurement of hardware, software, and services. Implementation faced challenges from the beginning: Verizon’s win was contested, delaying the start of work. SSA’s team didn’t realize the solution Verizon proposed, reinforced by SSA’s own contract requirements, was based on architectural components that were a generation behind leading contact center systems. NGTP’s 10-year planning horizon meant any solution would likely be obsolete before full deployment.
By 2020, with the project still in early development, the COVID-19 pandemic forced SSA call center agents to work remotely — a capability the existing legacy system lacked. Verizon scrambled to assemble a custom stopgap solution, but this was plagued with issues. From May 2021 to December 2022, over 40 service disruptions caused dropped calls, long wait times, and outages. At times, more than half of calls went unanswered as the team capped incoming calls to maintain system stability.
Meanwhile, NGTP suffered further delays and technical hurdles. SSA executives were frustrated but assumed they were contractually stuck. The system finally launched in December 2023 for the 800-number only, delivering just part of the promised functionality. But the system experienced ongoing performance issues, including increased wait times and disconnected or unanswered calls that hindered the agency’s ability to serve the public. On August 22, 2024, after only about 10 months of operation, SSA transitioned the 800-Number Network off the NGTP platform and moved to a different telephony solution. The NGTP project cost SSA over $160 million and was abandoned within a year of deployment, with the agency reverting to an alternative telephony platform.
The failure was not attributable to a single cause. Interviews and oversight findings point instead to a combination of over-specification, missing mission outcomes, weak accountability mechanisms, long planning horizons, and an acquisition structure that made adaptation difficult.
It is also important to recognize the scale and complexity of SSA’s operating environment. The agency’s service delivery depends on hundreds of interdependent systems, many of which encode decades of policy and operational logic. Modernization efforts must contend not only with outdated technology, but with deeply embedded business rules and integration dependencies that are not always fully visible at the outset. These conditions increase the difficulty of both specification and implementation, regardless of acquisition approach.
Specificity Did Not Produce Control
A central lesson of NGTP is that specificity in requirements does not necessarily translate into control over outcomes. The solicitation and technical requirements were extensive and highly prescriptive. They incorporated staff input but lacked sustained user-centered validation and focused heavily on defining technical components rather than the operational outcomes the system was intended to achieve. In several cases, the contract mandated architectural approaches that constrained flexibility and effectively locked the program into solutions already lagging prevailing commercial practice.
The NGTP contract required the development of significant custom telephony capabilities in a market where mature commercial Contact-Center-as-a-Service (CCaaS) platforms already existed. Custom software and hardware development inherently carries greater risk than configuring established commercial platforms: the first buyer bears the cost of defects, scaling problems, and design errors that mature products have already identified and resolved. As a result, the program assumed substantial technical risk without clear evidence that SSA’s mission required a bespoke system.
The decision to pursue a custom telephony architecture also introduced structural technical risks. The system was intended to function as a “single enterprise contact center” capable of routing calls across SSA’s national network. In practice, however, the implemented solution consisted of six separate contact centers operating as independent queues rather than a unified system. According to the SSA Office of Inspector General, this configuration prevented calls from being dynamically rerouted between queues, limited agents to answering calls from a single queue, and could disconnect calls when agents logged out of one queue even if capacity existed elsewhere in the system. These limitations increased wait times and created operational inefficiencies. Efforts to resolve the architectural mismatch led to the development of a custom routing “brain” intended to connect the six queues—effectively reinventing load-balancing technologies that have been widely used and commercially mature for decades. The need to retrofit this architecture required multiple contract modifications and created ongoing operational challenges. As one SSA leader later observed, “Some people on the project might have known that load balancers had been mature for 30 years, but managers weren’t listening to them.”
The contract’s prescriptive structure also undermined the flexibility typically associated with its contract vehicle. Although NGTP was structured as an IDIQ, the narrowly defined solution space meant that many necessary adjustments required formal work orders or contract modifications. In practice, the program combined the administrative rigidity of traditional contracting with the technical risk of custom system development.
The detailed specifications locked the implementation into many types of outdated architectural assumptions. For example, certain components were required to be compatible with an old, yet unspecified, version of Internet Explorer, a browser Microsoft formally retired in 2022 in favor of Microsoft Edge. Rapidly evolving technology environments can render highly specific requirements obsolete before systems are delivered. At the same time, the extensive technical detail did not fully address practical operational considerations, such as ensuring that existing SSA call center staff could easily access and use the system in their day-to-day workflows.
Missing Mission Outcomes
The NGTP case also illustrates the limits of operator-focused metrics. SSA understandably focused on call volume and the ability of the system to handle surges in demand. Previous infrastructure could “top out” during predictable spikes, such as cost-of-living adjustment periods. Capacity therefore became a central concern.
But throughput alone is not the same as service performance. For beneficiaries, the meaningful outcomes include how long it takes to reach a representative, whether the issue is resolved on the first contact, how many interactions are required, and how long it takes to complete a request. Those mission outcomes were not adequately embedded in the contract’s performance framework.
Metrics such as average speed of answer did not fully capture the user experience, particularly when calls were dropped, or handled initially by automated systems, or callbacks were counted in ways that reduced reported wait times without necessarily reducing the time required for beneficiaries to obtain help.
The deeper problem was architectural as well as contractual. SSA’s call center is best understood as a front-end interface to a much larger, deeply complex service delivery system involving eligibility determination, identity verification, claims processing, and payments. Yet the contract largely treated telephony modernization as a standalone technical problem rather than as part of an integrated operating model. This narrow framing also limited foresight into how the capability could evolve over time, adopting future emerging technologies or adding integrations with other agency systems to support an omnichannel service model. Defined primarily within a technical infrastructure context, the effort optimized for telephony components rather than positioning customer service as a strategic, cross-agency capability.
Accountability Was Weak Where It Mattered Most
Federal acquisition frameworks already provide multiple mechanisms for vendor accountability, including service level agreements (SLAs), financial incentives and penalties, option periods tied to demonstrated progress, and formal performance reviews. In the private sector, large IT and service contracts routinely embed such operational standards like uptime guarantees, response-time thresholds, incident-resolution timelines, and financial penalties for failure to meet them to ensure that vendors remain accountable for system performance under real operating conditions. In the NGTP case, however, these mechanisms were not sufficiently embedded in the contract structure or tied to mission outcomes and enforceable operational standards.
The SSA Office of Inspector General found that the NGTP contract lacked sufficient performance-based quality standards and incentives to ensure accountability for resolving system-performance issues. The practical result was limited leverage for the government even when the system failed to meet technical and operational needs.
The most striking example came at termination. When SSA stopped work on the NGTP effort, the agency still paid the vendor the remaining portion of the full $125M contract amount. Whatever the legal and operational considerations behind that decision, the message to the market was problematic: poor performance did not produce a proportionate financial consequence.
SSA’s Course Correction
SSA’s response illustrates an alternative approach. Rather than pursuing another large, fully specified replacement effort, the agency adopted a more incremental approach using cloud-native technology and more flexible contract mechanisms. A proof-of-concept deployment of Amazon Connect at a Pennsylvania call center allowed SSA to test the platform in live operating conditions before scaling further.
This approach introduced several disciplines that had been missing from NGTP. It reduced dependence on bespoke infrastructure, created an opportunity to measure performance under real conditions, and allowed the agency to collect operational evidence before broader rollout. Critically, assumptions were tested incrementally rather than embedded upfront. The agency also adopted Product Operating Model best practices: they stood up a cross-functional product team with a product manager, technical lead, design lead, and an SME lead who was responsible for state specific launches, training, and key metrics.
Early results suggested improvement. SSA’s Office of Inspector General reported that the agency’s telephone service handled substantially more callers in fiscal year 2025 and that reported average speed of answer improved. The subsequent administration leveraged the scalable platform to expand deployment across all field offices. At the same time, oversight and public reporting also highlighted the importance of careful metric design. Some reported gains did not fully reflect the total time beneficiaries waited for callbacks or to resolve their issues. That distinction is key: better performance frameworks depend not simply on more metrics, but on the right metrics.
Lessons for Outcome-Based Acquisition
The SSA case highlights several lessons:
- Complex systems cannot be fully specified in advance. Over-specification increases risk, and can lock programs into the wrong solution.
- Iterative delivery is a risk management tool. It surfaces integration, usability, security, and performance problems early enough to address them.
- Accountability must be tied to mission outcomes. Operational and customer experience results matter more than intermediate artifacts.
Governance matters as much as contract structure. Strong product ownership and leadership are essential. Critical to the successful turnaround was having a cross-functional “product quad” of product management, engineering, design, and domain expertise. In the NGTP case, requirements were largely defined within an infrastructure-oriented telecommunications function, leading to a solution optimized for technical components rather than end-to-end service outcomes. This organizational starting point constrained problem framing and limited the program’s ability to align delivery with user needs and mission performance.
An outcome-based model would have defined mission metrics such as first-contact resolution and total time to complete transactions, incorporated discovery phases, and tied continuation decisions to demonstrated performance. It also would have created a precedent for early adoption of critical monitoring tools used by leaders in the course correction, like integrating real-time customer experience telemetry into daily operations, which enabled continuous monitoring of user outcomes and rapid reprioritization of features to address emerging issues as they occur.
Finally, contract structure alone is not sufficient. Successful implementation depends on sustained leadership, technical judgment, and the institutional willingness to act on evidence. Several interviewees noted that meaningful progress accelerated only after leadership with prior agile and product delivery experience assumed responsibility for the effort. Acquisition structure can enable better outcomes, but it cannot substitute for leadership capable of making informed technical and operational decisions in complex environments.
Conclusion
Large-scale IT modernization is central to federal mission delivery. Traditional acquisition models remain effective in stable, well-defined environments but are poorly matched to software-intensive systems characterized by uncertainty, interdependence, and continuous change.
Outcome-based contracting provides a more effective framework for these conditions. It strengthens accountability by tying funding and continuation decisions to measurable performance, improves risk management through iterative delivery, and reorients acquisition toward public value. Rather than asking whether a contractor delivered what was specified, it asks whether the government achieved the mission results it needed.
Realizing this shift requires more than changes to contract structure. The authorities to pursue outcome-based approaches largely already exist, but incentives, funding constraints, and workforce capabilities continue to reinforce specification-driven models. Appropriations structures limit flexibility, oversight mechanisms emphasize compliance over performance, and many agencies lack the product management and data capabilities needed to define and act on outcome metrics. Addressing these constraints will require coordinated changes across budgeting, oversight, acquisition practice, and workforce development.
In the near term, IT modernization progress should be visible in concrete ways: contracts that tie option decisions and incentives to mission outcomes; programs operating with empowered Product Owners and real-time performance data; and evaluation frameworks that prioritize whether services are improving, not just whether requirements were met. Over time, this would mark a broader shift from managing compliance with plans to managing performance against outcomes.For technology and IT modernization efforts, the success of outcome-based contracting depends on alignment with product operating model practices, technical expertise, and sustained leadership. The central proposition of OBC is not less discipline, but better discipline—organized around measurable outcomes, empirical evidence, and the continuous identification and reduction of technical and operational risk.
Building Human Infrastructure to Mitigate AI Fairness Harms in K-12 Education
The rapid introduction of tools powered by artificial intelligence (AI) in K-12 education offers promises of data-driven personalized learning, real-time feedback, and relief for educators’ overstretched workloads. However, increasing access to emerging technologies alone is insufficient for achieving this vision. Without sustained, high-quality professional learning (PL), AI risks deepening a “digital design divide“— a gap where educators lack the support necessary to transform learning experiences by leveraging technology responsibly and effectively.
This challenge is not new. It mirrors a long-standing phenomenon in K-12 education where significant technology acquisitions occur without due efforts to sustainably build educator capacity. To mitigate this risk, state legislatures and education agencies must prioritize investments in human infrastructure– especially teachers, moving beyond systems that prioritize short-term tool training toward durable, high-quality professional learning systems.
Challenge and Opportunity
While a majority of U.S. educators now use AI in their work, the necessary support to use these tools effectively and responsibly lags significantly. According to RAND, half of the nations’ school districts have not provided training on AI, and high-poverty districts are even less likely to have provided training compared to their low-poverty counterparts. The failure to provide this essential support and the resulting disparity poses a dual fairness risk for vulnerable student groups. They may be subjected to biased or harmful AI practices, and they are also more likely to miss out on the innovative uses of AI, including deeply personalized learning responsive to their strengths, backgrounds, experiences, prior knowledge, and needs.
Furthermore, recent research identifies four systemic issues in current systems that govern professional learning (PL) for high-quality, technology-enabled instruction:
- Inconsistent Definitions: While K-12 leaders share a vision for student-centered instruction where technology like AI deepens and accelerates learning, few have a formal definition of what this looks like in practice or how PL supports it.
- Short-Term Funding Defaults: Funding often targets “one-off” workshops for specific tools and platforms. These meet immediate rollout needs but fail to build durable educator capacity in a rapidly evolving landscape.
- Compliance-Driven Monitoring: Tracking the impact of PL efforts often focuses on box-checking for relevant laws and regulations, rather than using data to refine instructional strategy.
- Lack of Documented Models: State leaders struggle to synthesize and disseminate locally-initiated, well-documented PL programs with evidence of success.
The real opportunity of AI lies not just in the tools, but in an educator workforce prepared to wield them. High-quality PL must thus move beyond short-term tool training to focus on areas necessary for equitable implementation, such as AI fairness and bias mitigation, ethical use of data, critical thinking, data foundations, and deep integration of AI-enabled tools into standards-aligned, high-quality instruction. When done right, this investment in human infrastructure ensures AI accelerates learning outcomes for all students, closing the “digital design divide.”
State legislatures and education agencies are pivotal actors who must address this issue through strategic policy levers. While individual districts manage much of the budget implementation and programmatic decisions, states set the conditions for local success by aligning funding streams and defining clear instructional visions.
Plan of Action
Recommendation 1. Define and Promote Aligned Visions of AI-Enabled Instruction
- Establish Definitions: Led by the state education agency and developed with broad input from stakeholders (e.g., educators, students, families, industry, research), publish and embed a statewide definition of high-quality, AI-enabled instruction in existing guidance and policy. For instance, states have leveraged frameworks like the ISTE Standards, CSTA Learning Priorities, and Universal Design for Learning guidelines to inform their definitions.
- Align Funding Streams: Require districts applying for PL funds, including the $2 billion Title II-A program under the Elementary and Secondary Education Act (ESEA), to describe how their proposed activities align with the state’s articulated vision.
- Provide Exemplars: Curate sample classroom artifacts and PL tools that reflect the vision.
Recommendation 2. Align Funding With Instructional Priorities
- Name Allowable Uses: Explicitly list AI and digital learning supports as allowable uses in guidance for various PL funding streams.
- Ensure Deliberate Procurement: Develop guidelines that encourage the procurement of safe, evidence-based, inclusive, usable, and interoperable tools, as well as risk and impact assessments for AI-powered edtech.
- Synchronize Cycles: Align grant cycles and reporting deadlines so districts can plan coherent, multi-year PL investments.
Recommendation 3. Leverage Compliance Structures for Continuous Improvement
- Shift Monitoring Metrics: Create systems that prompt districts to report not only educator attendance in PL efforts, but also shifts in practice and student impact, with a focus on non-punitive, streamlined data collection to minimize administrative burden.
- Collaborative Coaching: Use monitoring meetings as coaching sessions in addition to compliance audits, providing clear, actionable feedback to improve PL activities.
Recommendation 4. Encourage Durable Professional Learning Models
- Prioritize Sustainable Roles: Structure funding to prioritize ongoing coaching networks and dedicated AI readiness specialists over standalone training.
- Regional Infrastructure: Partner with regional education service centers to provide shared coaching staff across multiple districts.
- Elevate Educator Voice: Encourage PL programs establish formal mechanisms for teacher input in design, iteration, and evaluation to ensure practical relevance and quality.
Recommendation 5. Work Across Silos in State Leadership
- Cross-Departmental Collaboration: Establish regular touchpoints between key staff—specifically federal programs directors (e.g., ESEA Title program leads), curriculum leads, and edtech leaders—to align priorities and ensure funding streams work in concert.
- Joint Guidance and Pilots: Publish guidance that connects AI literacy directly to curriculum standards and assessment strategies. Launch pilot projects that combine resources from multiple departments, such as blending AI literacy PL with new project-based learning initiatives, and evaluate them together.
Recommendation 6. Document, Highlight, and Scale What Works
- State Repositories: Launch an online hub or in-person showcases where districts can share artifacts of model PL activities and road maps for implementing effective coaching initiatives.
- Responsible Innovation Amplification: Create awards or public recognition programs that spotlight measurable progress in AI-enabled instruction, helping to inspire others.
State education agencies specifically can adapt these recommendations based on their current capacity and context. For example:
- Low-Intensity
- Explicitly note across existing guidance documents (e.g., ESEA Title program funding guidance) that federal and state funds can support PL tied to AI-enabled instruction.
- Curate aligned PL examples to model high-quality investments into human infrastructure.
- Medium-Intensity
- Publish a statewide vision for AI-enabled instruction and responsible AI use, and integrate this vision into grantmaking processes.
- Convene alignment workshops and develop sample braided budgets that connect various streams of funding.
- Create and disseminate a high-quality PL model on AI fairness and use for optional adoption by districts, encouraging delivery through local or regional coaching networks for maximum relevance and sustainability
- High-Intensity
- Codify durable PL structures in policy, including by embedding roles like instructional technology coaches in state regulations or quality standards, prioritizing the development of internal educator capacity and clear teacher-to-coach career pathways.
- Institutionalize cross-departmental collaboration by establishing offices or leadership teams that coordinate curriculum, assessment, technology, and professional learning.
Conclusion
According to SETDA’s edtech trends survey, AI is currently the leading state edtech priority and top state initiative. However, with only a small group of states currently prioritizing existing funds for technology training, there is an immediate need to improve the systems governing professional learning. By investing in the “human infrastructure,” as exemplified by states like Wyoming and Massachusetts, state leaders can ensure that AI becomes a tool for accelerating outcomes for all students.
Sustaining Scientific Collections in the Age of AI
Scientific collections play an important but underappreciated role in the American science and technology ecosystem. Their existence has accelerated science by providing repositories for samples, archived information and data. This increases the efficiency of scientific research by mitigating the need to repeat field work while serving as essential information libraries. Collections are particularly important for the study of biological specimens and pathogens, offering significant advantages to human health and food security, but also extend to the physical and geological sciences, reducing the need to develop new observations. The decentralized nature of scientific collections–of which there are more than 10,000 globally and about 2,500 in the United States–increases the value of digital curation technologies and particularly those supported by machine learning and artificial intelligence.
AI companies have identified that one of the most exciting value propositions for their models is the potential acceleration of science by rapidly synthesizing information from disparate sources and gleaning new and undiscovered insights. Accomplishing this mission inherently requires access to public goods and resources, and (in particular) training information that is relevant for scientific discovery. We believe there is a compelling case to be made for AI companies to contribute to the sustainability of that public good, whether it be through direct financial support or through the provision of in-kind tools that improve the management and sustainability of these uniquely important sources of training information.
A scientific collection is typically defined as an organized, curated repository of physical or digital specimens that (a) support scientific research, (b) are maintained according to systematic standards, and (c) enable reuse or reanalysis. Artificial intelligence models seeking to accelerate scientific discovery require stable, curated and maintained sources from which to train and draw information, and are already trained using scientific collections (in addition to other, lower quality data sources). AlphaFold’s Nobel-prize winning efforts, for instance, were supported by information derived from the Protein Data Bank, carefully collected and maintained over decades of scientific experiments.
While operating collections is relatively inexpensive (with notable exceptions, like biobanks), their position in markets is challenging as the products created by the “goods” in scientific collections tend to be made open and available for the broader scientific community. As a result, collections are often treated by their owners as a non-revenue-generating institutional expense. In order to foster the economic viability of collections while providing accurate outputs that are valuable to the scientific community, collections managers should update their terms of use and require that AI companies play the role of responsible stewards of scientific data and information, consistent with the role other companies play in supporting successful American research infrastructure.
The Cooperative Stewardship Model for research infrastructure, which has served as a reliable template for balancing openness and utilization with commercial interests, offers a possible pathway for collections’ sustainment. Federal agencies should seek to promote high-quality processed information and primary sources wherever possible, including through grantmaking, contracts, and procurement. Given that the primary method by which the general public, and particularly students, engage with scientific information is already mediated by AI (through search engines or otherwise), the trustworthiness of AI platforms and responsible stewardship of collections as a primary data source is paramount.
Challenge and Opportunity
Viability of Collections
Scientific collections are one of the most critical elements of the research ecosystem, supporting everything from food security, like the National Seed Storage Laboratory in Fort Collins, to the Smithsonian’s mosquito collections which enable research on zoonotic pathogens. To adopt the language of the tech world, these are “full stack” research endeavors requiring specialized knowledge, capabilities, and environments to do successfully. Yet despite their relatively low costs, in recent years major scientific collections, like the herbarium at Duke University, have faced significant maintenance and upkeep challenges due to a lack of revenue sources. The challenges and viability of specific collections can vary dramatically due to the fact that collections represent a form of extremely decentralized infrastructure for financial, cultural, and legal reasons. For instance, the operation of the Smithsonian National Museum of Natural History’s collections may be substantially different than privately-owned collections of insects or those representing artifacts, samples, or libraries stewarded by Indigenous Peoples.
Recent federal policy actions, particularly with respect to the introduction of new constraints on indirect costs, threatens to further undermine the business model of collections and force the loss or closure of vital sources of scientific data and information. Collections, which are relatively inexpensive to maintain relative to other scientific infrastructure, still require a significant number of programmatic activities to curate, maintain, and support at a scale similar to other research activities. An example of the types of costs, which can vary widely based on the size of the collection and what is included, associated with collections activities are provided, below:
The cooperative stewardship model for research and development infrastructure has provided the backbone for federal scientific collections for many decades. While most infrastructure is free to use and accessed for scientific purposes, for-profit enterprises are generally charged for access to infrastructure in order to sustain, maintain, and provide new capabilities relevant for industry. Examples include the National Institute of Standards and Technology’s repository of standard reference materials, widely used by industry, which includes everything from $1200 peanut butter reference material to standard cigarettes for ignition resistance testing. Major U.S. scientific facilities generally operate on the basis of full cost recovery for services used by commercial providers as opposed to relying on those providers as a primary source of revenue generation. For collections, this would help reduce pressure or concerns stemming from political pressure on indirect costs from grants.
The AI companies and their leaders have identified science as one of their most exciting areas for development, and suggested new licensing and revenue models for measurable AI outputs. For sure, models have the potential to serve as tools for scientific discovery by providing quick access to massive volumes of information largely derived from both scientific literature and data sources and allowing scientists to rapidly extract novel insights from that information. It is reasonable to imagine a scenario where the primary interaction that scientists have with collections in the future is mediated by training data fed into AI foundation models.
AI models must accurately reflect information from scientific collections
AI model firms, which rely on quality sources of training data, ideally should have an express interest in seeing reliable scientific information sources maintained and managed in a way that improves the performance of models due to the availability of quality data. Good information sources, like collections, must be available and maintained if companies are going to successfully implement the vision of AI for science expressed by their marketing and executives. It requires taking information derived from the physical world, digitizing it in a way that is comprehensible to a computer, and then feeding that data and relevant processed information into an AI model where it may be used to help with the process of scientific discovery.
Some of these data sources are quite large and processing-intensive. For instance, the world’s largest radio wave telescope, the Square Kilometer Array based out of South Africa, is expected to generate approximately 700 petabytes of astronomical data every year–about seven times more than that which is generated by Google Search in the same time period. Models seeking to leverage these data sources for discovery will likely place additional and significant burdens on scientific data infrastructure.
Current AI models are not always the most reliable sources of scientific information, in part due to frequent “hallucinations.” These models do not always preserve scientific information with fidelity, even if trained on high-quality data. For instance, when asked to produce an image of an anatomically correct blue whale skeleton, OpenAI’s ChatGPT model will output images of a strangely toothed (as opposed to baleen) whale skeleton that incorrectly represents the whale’s tail as a single, large bony structure, among other errors. Search engines have similar issues, with regular Google searches in late 2025 producing a humpback whale image when searching for “blue whale,” citing a popular science website as opposed to a scientific collection or other reliable source.

Image generated by ChatGPT using the prompt “Can you draw me an anatomically correct image of a blue whale skeleton?” on April 2, 2026. Note the teeth-like structures on a baleen whale, the bizarre pectoral fin bones, and the hallucination of a bone representing the tail, among other errors. Also note the lack of a clear watermark identifying the image as AI-generated.
Google search result for “blue whale,” November 12, 2025. Note that the first image, drawn from ZME Science, is actually a photograph of a humpback whale.
The challenge of producing high quality scientific outputs does not (and should not) rest entirely at the feet of model developers. While these data sources provide us with the most complete library of field samples, they are far from perfect, oftentimes tagged with partial, non-standard, and more infrequently incorrect information (for instance, samples collected before 1990 generally are not tagged with latitude and longitude). This makes it more difficult to judge the quality of the inputs, requires human curation, and occasionally additional field work to improve the quality of the collection. This actually presents an opportunity for AI models in science, which could (with appropriate validation) help identify and correct missing, incomplete, or inaccurate information.
There is a general need for improved transparency with respect to training data and the information standards models use to evaluate the quality of data.
Scientific collections are uniquely valuable to foundation models given the high quality of the data and information sources, as distinct from information sourced from the broader internet. Unfortunately, it is difficult to assess whether companies that develop generalized models, like OpenAI or Anthropic, privilege scientific data sources over general information collected from the internet, which may include creative renderings, fantastical artist depictions, and outright falsehoods. Assessing this challenge is not straightforward. Companies frequently do not disclose training information or details about how their models process data and information–if they are even able to explain a model’s output.
Recommendation 1. Scientific collections should update their terms of use to require that data used to train for-profit models be licensed, consistent with existing principles for research and development infrastructure.
Scientific collections, many of which are already struggling, are now faced with an existential threat as a result of the government’s budget cuts and federal policy actions (particularly those related to indirect costs). Modifications to collections’ business models are already inevitable, especially as industry and philanthropy are rapidly reprioritizing their current investments. Rather than attempting to create a new relationship between these companies and institutions that provide this valuable infrastructure, it makes sense to rely on proven public-private partnership infrastructure governance models identified in the National Academies’ cooperative stewardship report, rather than building a new social contract that might threaten the general accessibility (for both AI companies and the broader scientific community) of this critical source of information.
Collections offer a unique and necessary service if artificial intelligence is to realize its full potential for scientific discovery. Rather than attempting to develop or adopt new and more unfamiliar business models, collections should adopt the same cooperative stewardship principles that work for the remainder of the science and technology ecosystem. There is inherent value in making sure that collections serve as the primary basis for information used to train AI models for scientific discovery, as opposed to other sources of information that might be pulled from less reputable sources in the broader internet (including scientific studies that resist replication).
Companies that wish to use the products of research for proprietary and commercial purposes are similarly charged for access to infrastructure. Organizations seeking to produce non-proprietary models for scientific purposes, similarly, should be given the same access advantages conferred to other parts of the scientific community to ensure dissemination and broad utilization.
It is reasonable to assume that AI companies could respond to fee-for-access by simply not accessing data or information from collections. Efforts to derive information from papers or other sources are likely to be incomplete and less useful for the creation of high-quality models useful to science. It is reasonable to ask–as is the case for every other industry that utilizes R&D infrastructure–for companies that stand to profit from the products of collections to pay for their sustainment.
Alternatives, including the provision of in-kind tools or resources to assist with the curation and management of scientific collections (and thereby increasing the efficiency of collections and improving the quality of the services they provide the broader community), may also be explored.
Recommendation 2. Science agencies should prioritize development of specialized tools to curate information and data housed in scientific collections and assist in the development of data and information standards. These tools should continue to rely on cooperative stewardship principles (i.e.,allowing open access for non-commercial use while maintaining fee-based access for profit-seeking entities).
Given that today’s generalized LLMs, image generators, and search engines cannot be counted on to reliably output accurate scientific information, science agencies should continue to invest in specialized models that are designed and developed to address critical mission needs. This would likely require investment in models that are designed to curate, compare, and manage samples which are frequently tagged with non-standardized or lower-quality information.
Recommendation 3. The government should show preference when procuring solutions for science and technology purposes for AI models that are trained on data and information in scientific collections. This should include investment in R&D to help develop models that appropriately privilege high-quality training information.
Government procurement can provide an important tool for increasing the quality of services through the procurement process. As government agencies invest in artificial intelligence for policy purposes, including for the development of technical tools that affect people’s rights and safety, the need for models to output quality information increases substantially.
Government agencies can, through the contracting process, request that providers be held to certain standards and, whenever possible, provide appropriately-licensed images drawn from primary sources. Such requirements could also substantially reduce the compute requirements produced by models, which could simply offer a reference image, video, or other information source as opposed to generating (and possibly hallucinating) a lower-quality output produced by the model itself.
Conclusion
The 2020 Economic Analyses of Federal Scientific Collections produced by the Smithsonian and National Science and Technology Council notes the conception of collections by some authors as a “global public good” underpinning much of the science and technology enterprise. As scientists’ and the publics’ interactions with collections and the knowledge contained within them becomes primarily mediated by AI platforms, and as AI platforms seek to profit from content created from their use, the role of those platforms as stewards of these resources should correspondingly increase. Collections are ideal “senses” through which AI platforms can collect high quality information about the world, enabling the most exciting implementations of AI to be realized. Supporting the infrastructure that enables that realization is crucial for the success of the technology.
Thank you to Dr. Oliver Stephenson for his contributions to the text.
A People-centered, Power-conscious Regulatory Democracy Balancing Distributive Justice and Delivery Efficacy
Building Blocks to Make Solutions Stick
People-centered, power-conscious rulemaking, using deliberate stakeholder engagement strategies, produces faster and better results.
Implications for democratic governance:
- Stakeholder engagement strategies must understand and design for power disparities.
- Public participation in policy processes should be non-performative, while also avoiding meaninglessly chasing consensus.
- Legitimacy through visible reciprocity – show the public how their input shapes choices.
Capacity needs:
- Stop treating engagement as read only/listen only; set participation expectations to respond, ask, shape, and negotiate.
- Build the institutional muscle to make stakeholder engagement a light lift (e.g., through reusable engagement templates and tools).
- Enable sequential and and tailored participation, and incentivize early steers that flag risk and opportunity.
- Train staff in facilitation, organizing, translation, conflict navigation, and other engagement skills or hire staff that can bring these skills.
One of the biggest obstacles confronting meaningful action to address climate action are power disparities. Among our governing institutions, the federal administrative state is unique in its potential for overcoming these power disparities by offering an effective mechanism for redistributing political power from corporate interests committed to maintaining the status quo to the general public who are already bearing the costs of global climate disruption. The key to realizing this potential is “regulatory democracy.”
At present, though, the means for conducting public engagement in the administrative state generally fail to meaningfully engage the public, but instead have the perverse effect of reinforcing power disparities, as has been ably documented by the burgeoning Abundance movement. What is needed, instead, is a better approach to regulatory democracy – one that is people-centered and power-conscious.
This paper sketches out what a people-centered and power-conscious approach for improving regulatory democracy in the rulemaking process: a reform called “Public Participation Planning.” This reform consists of two major procedural components. First, it calls upon agencies to develop “public engagement strategy blueprints” as a mechanism for deliberately creating a tailored public engagement strategy for each rulemaking. The key innovation here is a recognition that different kinds of “expertise” (democratic vs. technocratic) are required at different stages of a regulatory development – a concept referred to here as “sequential participation.” Second, it calls upon agencies to document the actual performance of that strategy, by including a group of documents called the Initial and Final Public Participation Plan Statements. These statements would capture the impacts that the public engagement actions have on the development of the rule. They would be included in the rulemaking document along with the notice of proposed rulemaking and final rules, respectively, where they can help inform judicial review of the rule. In theory, a president could implement a rigorous version of Public Participation Planning without new statutory authority. Still, the full potential of this reform could be enhanced with additional actions by Congress and the judiciary. Properly implemented, Public Participation Planning would both improve the quality of agency decision-making and permit for more expeditious policy implementation by reducing the ability of powerful interests to use the rulemaking process to reinforce a bias toward maintaining the status quo.
Addressing the Climate Crisis Through Better Regulatory Democracy
One of the underappreciated features of the federal administrative state – at least within our contemporary context – is its potential capacity to prevent politically and economically destabilizing concentrations of power from taking hold by continually redistributing it to the general public. That the architects of the modern administrative state – turn-of-the-20th century policymakers, thinkers, and movement leaders alike – designed it with this particular goal in mind seems to have been lost to history, however.
The key to their radical vision of the administrative state was “regulatory democracy” – that is, the notion that administrative agencies would work cooperatively (and sometimes competitively) with the public to shape policy priorities, design, and implementation. At its best, regulatory democracy would take the form of a working relationship that was ongoing and durable. The dynamic it was meant to yield would be a much thicker form of engagement in our governing institutions than ordinary Americans would experience through the episodic opportunities of casting a ballot and the often-binary choices they would be presented with during those opportunities.
This vision should be particularly resonant today, though it may sound esoteric and peripheral at first blush. For the reformers of the early progressive era, creating a new venue for translating public power into policy change was essential for effectively meeting the then-emerging challenges that many Americans faced due to such societal changes as industrialization and urbanization. Today, we face cascading challenges – climate change, globalization, and rapidly evolving forms of computational technology such as Artificial Intelligence and quantum computing – which have similarly exposed the limits of our governing institutions. Then, as now, society was characterized by vast disparities of economic and political power that further threatened effective policy implementation. The administrative state’s comparatively decentralized and democratized design – relative to Congress – was meant to mitigate these effects.
More to the point, if we are to avert the worst consequences of the climate crisis, we will need to quickly revive these robust democratic traditions of the administrative state. After all, as the Green New Deal movement correctly taught us, power disparities are a root cause of this crisis; effectively decarbonizing our economy and investing in infrastructure that is hardened to withstand the unavoidable impacts of climate change will require confronting these same power disparities. Among our governing institutions, the administrative state is best equipped to meet this kind of challenge under these kinds of circumstances.
Achieving the full democratic potential of the administrative state will require some important reforms, however. As the primary law governing the operations of the administrative state, the Administrative Procedure Act (APA) establishes many of the mechanisms that agencies use for democratic engagement. For informal rulemaking, which has become a leading vehicle for administrative policymaking, the APA creates the notice-and-comment procedures. The benefit of decades of experience has revealed that these procedures not only fail to meaningfully engage large segments of the population; they also reinforce status quo inaction and the underlying power disparities that benefit from such inaction.
This white paper argues that what is needed instead is an approach to public engagement that distinguishes between the different kinds of stakeholders implicated by a given policy action and accounts for the underlying power disparities that define their relationships to the policy problem that the action is intended to solve. As discussed below, the passive, power-agnostic posture of notice and comment fails to accomplish either of these objectives. Among other things, a people-centered, power-conscious approach would require carefully cataloging the universe of relevant stakeholders, the barriers they face to meaningful engagement, and the kind of input those individuals would likely bring to inform a policy decision.
A people-centered, power-conscious regulatory democracy would also require deliberate attention to how best to obtain public input. It would demand that agencies have a ready toolbox of engagement tactics and solutions tailored to effectively obtain different kinds of input from different kinds of stakeholders. It would also require agencies to plot out in advance the different stages in their rule development process for deploying those tactics and solutions – a concept this white paper refers to as sequential participation.
This approach would depend for its success upon a sincere commitment to transparency and reciprocity with stakeholders. Agencies would need to continually communicate with stakeholders and be completely forthright in those communications – even when the news is not what those stakeholders will want to hear. They will also need to carefully document how public engagement was conducted throughout the rulemaking process and the role it had, if any, on the progression of decision-making at the different stages of that process.
Lastly, and perhaps most controversially, this approach should draw on the agonistic model of democracy. Practically speaking, that means the goal of regulatory democracy should be to surface and channel productive disagreement, rather than embark on a quixotic search for consensus or near-consensus on controversial policy matters. As explained below, powerful interests have used consensus-based approaches to decision-making as a kind of veto-gate to defend their preference for the status quo. Reorienting our expectations for regulatory democracy in this manner will thus permit meaningful engagement without unduly sacrificing timely policy implementation – a concern that has achieved greater prominence due to the Abundance movement.
These lessons could, of course, just as readily apply to reforming state- and local-level administrative procedures as well. Indeed, subnational governments are already playing a pivotal role in addressing the climate crisis, particularly while steadfast Republican obstruction has left Congress incapacitated on this issue. The state rulemaking procedures or public utility commission proceedings that are responsible for implementing these policies could be strengthened through a people-centered, power-conscious regulatory democracy program. For simplicity, however, this white paper will focus on developing a version of this approach that applies to the federal rulemaking process.
While there may be several different methods for institutionalizing a people-centered, power-conscious regulatory democracy, this white paper proposes the use of what it calls Public Participation Planning. Under this reform, agencies would develop tailored plans called public engagement strategy blueprints. The purpose of these blueprints is to ensure meaningful engagement by relevant stakeholder groups – especially those representing communities that are structurally marginalized or that historically have been excluded from democratic processes. Critically, these blueprints would account for the entire rulemaking process with the aim of proactively engaging particular stakeholders at stages where their input is most likely to be relevant and useful. This proposal would also call on agencies to document their outreach and engagement actions and any impacts they had on the proposed and final rule in a special report called a Public Participation Planning Statement, which would be made part of the rulemaking record.
The practical advantage of Public Participation Planning is that it could be instituted by a president with existing legal authority. Still, the proposal also outlines how the other federal governing institutions – including Congress and the judiciary – can help ensure that the benefits of public participation plans achieve their full potential. One important task for the coordinate branches would be to address whether and to what extent existing administrative law doctrines, such as Vermont Yankee, present barriers to achieving the full potential of Public Participation Planning for advancing regulatory democracy. It is also worth emphasizing that parallel efforts to reinvigorate the most important democratic institution in our constitutional framework – Congress – will also be necessary to realize the full potential of regulatory democracy. After all, the administrative state can only implement the laws that Congress passes. It will thus be more effective for the administrative state to leverage regulatory democracy to tackle something like the climate crisis if Congress were to pass legislation explicitly directed at that issue.
If properly implemented, a comprehensive reform program to accomplish regulatory democracy that is people-centered and power-conscious could be essential for addressing complex policy changes such as the climate challenge. As Public Participation Planning demonstrates, this approach would both improve the quality of agency decision-making and permit expeditious policy implementation.
Background: Regulatory Democracy at a Crossroads
Regulatory democracy – represented not just by the APA’s notice-and-comment procedures but also the National Environmental Policy Act’s analytical requirements and their state– and local-level analogs – has come under increasing criticism from several directions in recent decades. Concerns that the regulatory system is undermined by too much public participation stretch back to at least the Obama administration. At the behest of Cass Sunstein, then Office of Information and Regulatory Affairs (OIRA) Administrator, agencies during this era strongly embraced cost-benefit analysis and other technocratic decision-making tools as an apparent antidote to the irrationality and “mistakes” that ordinary lay people make. Under this approach, public participation was to be viewed with extreme skepticism – if not outright hostility – and thus minimized as much as possible.
More recently, the Abundance movement that has emerged in recent years has come to single out for criticism many of the existing public participation requirements in administrative law. According to this criticism, powerful entities often abuse such requirements as a means for delaying policies that they oppose. Conservatives have also begun to reject public participation, with President Trump having directed agencies to evade notice-and-comment procedures whenever possible during this second term. This move seems in keeping with his overall crusade to centralize administrative power in the White House and build something akin to an authoritarian administrative state.
At the same time, we have seen a growing movement among policymakers and advocates focused on expanding public participation opportunities. Notably, as part of his administration’s larger Modernizing Regulatory Review project, President Biden issued a memorandum providing agencies with guidance on how to strengthen public engagement in the rulemaking process – with a particular focus on marginalized communities. Among other things, this memo encouraged agencies to deploy various strategies for engaging members of these communities at the earliest stages of the rulemaking process. Separately, a group of progressive members of Congress have promoted a comprehensive regulatory reform bill called the EXPERTS Act. One of its provisions would create the Office of Public Advocate, which would be charged with supporting individuals and other underrepresented groups in the notice-and-comment process.
What these competing movements reveal is that almost no one is satisfied with the current approaches to regulatory democracy. This dissatisfaction, in turn, arises from both practical flaws and theoretical disagreements associated with these current approaches.
Practical Flaws of Regulatory Democracy
Due to poor design, the prevailing approaches to regulatory democracy generally fail to effectively engage most members of the public in critical administrative state tasks of decisionmaking and implementation. Worse still, in many cases, these design flaws can combine in ways that function to systematically exclude members of many structurally marginalized communities, thereby reinforcing the very power disparities that are often at the root of the policy problems that regulations are often designed to address.
Many of these approaches follow a rigid, one-size-fits-all design that prevents their implementation from being adapted to meet the unique demands that can arise in different policymaking contexts. This can be seen in the APA’s informal rulemaking context. The same basic notice-and-comment process applies for policies as varied as setting Medicaid reimbursement rates and regulating the use of non-compete clauses in employee contracts. Yet, each of these policymaking contexts involve very different kinds of stakeholders whose relationships are characterized by different kinds of power structures. As such, effectively obtaining input from these different kinds of stakeholders is likely to require different, tailored engagement tactics.
To be sure, the procedural requirements setting out the public participation mechanisms set a legal floor; a president generally has the authority under Article II of the Constitution to go above and beyond by adding new public participation strategies aimed at alleviating the flaws that arise from the mandatory approaches. Indeed, as noted above, the Biden administration undertook some steps along these lines. In general, presidents are unlikely to undertake such steps without a clear strategy for doing so due to budget constraints and the growing criticism of public participation in the regulatory system noted above.
One important adjustment a future president could take to help make regulatory democracy opportunities more inclusive for structurally marginalized communities is to introduce them earlier in the policy development process. Presently, most public engagement opportunities tend to occur relatively late in policy development, as the APA notice-and-comment process illustrates. By this point in regulatory development, many of the foundational decisions leading up to the regulatory proposal have been resolved, including problem definition and solution scoping (not to mention the decision to prioritize this particular rulemaking at all). The remaining issues left open for public input are the kind of esoteric or technical details that are typically well beyond the knowledge or expertise of ordinary people.
Put differently, the manner in which regulatory democracy is currently conducted fails to account for the “sequential logic” of the policymaking process – a logic that necessarily draws on different kinds of expertise at different steps. An agency’s authorizing legislation, of course, sets the key parameters for what regulatory actions an agency might undertake and how it might design those actions. From there, though, important decisions remain such as which “public problems” are worthy of priority attention and how best to begin constructing the scope of policy solutions to meet those problems. These earliest stages in the policy development process call for a more democratic form of expertise that finds its source in stakeholder’s lived experience and situated knowledge. This kind of input might, for instance, spur the EPA to prioritize tackling pollution from industrialized agriculture or determine how stringently the Consumer Financial Protection Bureau regulates the use of forced arbitration clauses in consumer contracts.
In contrast, the more conventional understanding of technocratic expertise tends to become more relevant at later stages, such as when decision-makers must refine policies to account for such complex questions as applicable legal constraints, economic factors, the state of technology, or the body’s toxicological mechanisms. These issues might include what control equipment should be required to limit emissions of a toxic air pollutant or the potential energy use impacts of strengthened appliance efficiency standards. Not incidentally, social philosopher and an early intellectual force behind the modern administrative state John Dewey memorably captured the ordinal nature of the policymaking process with his observation that “the man who wears the shoe knows best that it pinches and where it pinches, even if the expert shoemaker is the best judge of how the trouble is to be remedied.”
The upshot of this failure is that the regulatory democracy currently privileges the kind of technocratic input that well-resourced interests committed to maintaining the status quo are uniquely well positioned to provide. Those who lack access to this expertise – including the resources and training to obtain it – are thus effectively prevented from meaningful participation.
Another important adjustment that agencies could make to improve public engagement is to conduct more affirmative outreach to specific stakeholders. Instead, current regulatory democracy approaches adhere to an “open door” model by which agencies invite input on equal terms from all interested stakeholders and then passively wait to receive whatever input is provided. The notice-and-comment procedures, of course, best illustrate this model, and it has been replicated in other regulatory forums as well, such as the “lobbying meetings” during OIRA’s centralized regulatory review process. At best, this model favors stakeholders with the resources to monitor and answer open door invitations for participation. In contrast, individuals and smaller community-based organizations are unlikely to consult the Federal Register on a daily basis or to have the technical capacity to parse a large rulemaking proposal to identify whether and how it implicates their unique interests.
In the worst cases, entrenched interests can abuse the open-door model by leveraging their vast superior resources to excessively voluminous comments containing information of only marginal utility or relevance – a practice known as “packing the record.” As legal scholar Wendy Wagner noted, it is not uncommon for industry interests to submit hundreds of pages worth of highly technical comments, resulting in rulemaking records that are more than 10,000 pages in length. These stakeholders engage in this kind of gamesmanship because they treat the notice-and-comment process more as a prelude to litigation over the final rule rather than as good faith attempt to improve the quality of the rule. Significantly, from a power perspective, this record-packing scheme only works if the party engaged is able to back it up with a credible threat of bringing litigation – something individuals and community-based organizations are unable to do.
In effect, this gamesmanship has enabled powerful interests to install the courts as the primary locus of regulatory decision-making, on the apparent presumption that they will afford a more sympathetic audience for their policy arguments – usually in favor of maintaining the status quo – than what they may find at the agencies. This tactic has the additional benefit of contributing to regulatory ossification, as agencies seek to “bulletproof” their rules as much as possible to avoid adverse results on subsequent judicial review. The advent of Artificial Intelligence suggests that this problem could grow even worse in the future.
Significantly, various studies on the practice of regulatory democracy have documented the vast quantitative and qualitative disparities in participation that exists between unaffiliated individuals and organized interest groups. Empirical research on the APA’s notice-and-comment process in particular seems to confirm that the design of those procedures has the effect of systematically excluding most members of the public, particularly those from structurally marginalized communities. These results suggest that the notice-and-comment process is failing at its purported task of assembling something approximating comprehensive policy-relevant information for agency decision-makers by systematically depriving them of critical forms of “expertise” that tend to be in the exclusive possession of the individuals and communities who live closest to the problems that the policies are meant to solve. They are left to fix shoes without knowing where exactly they pinch their wearers.
The results of these studies also suggest that these kinds of shortcomings in the notice-and-comment process do not merely limit effective engagement; they also serve to exacerbate underlying power disparities. That is because the skewed perspective that agency decision-makers obtain through these procedures necessarily favors entrenched sites of political or economic power. In terms of substantive results, the public input obtained through the notice-and-comment process often translate into a strong status quo bias toward inaction – or, at best, towards actions that only minimally inconvenience empowered stakeholders. Thus, for example, the public comments for a rulemaking to address the climate crisis are likely to be dominated by fossil fuel interests – as opposed to those who are disproportionately harmed. To the extent that this public input creates a skewed picture for agencies of the harms that significant disruption to our climate systems will create, it risks militating against the kind of aggressive climate policies needed to avert these harms.
Lack of a Coherent Theoretical Basis for Regulatory Democracy
One of the major sticking points is that students of the administrative state have never really achieved something like a real universal agreement on why regulatory democracy is important in the first place. The business community and conservatives have questioned the legitimacy of the modern administrative state within the U.S. tripartite constitutional governing framework at least since the advent of the Great Society programs. In response, scholars have invoked a variety of democratic theories to salvage the constitutional legitimacy of the administrative state. (Though, for the most ardent of conservative critics, such as Philip Hamburger, no theory is likely to suffice.) In turn, the lack of a clear theoretical basis has hampered efforts to design effective public participation mechanisms – contributing to many of the practical flaws described above.
According to the pluralist model, the administrative state’s democratic features provided a forum in which competing interests could shape policy. As long as the forum provides a reasonably fair opportunity for engagement for all interested stakeholders, this theory assumes that the substantive results that emerge roughly approximate the common good.
Another theory is the civic republican model. Unlike the pluralist model, which holds that the administrative state becomes imbued with democratic legitimacy through the balancing of competing interests, civic republicanism starts from the position that some idealized notion of the common good exists externally from the administrative state. Revealing this concept of the common good – which, presumably, all stakeholders would recognize as worthy of their consent – can only be achieved through a process of careful reason-giving and deliberation. The administrative state’s democratic legitimacy thus hinges on its ability to enable such a process to take place.
The influence of these competing schools of thought can be seen in institutional and legal reforms over the last several decades. For instance, the Regulatory Flexibility Act’s procedural requirements aimed at ensuring that regulators account for small business concerns reflects the pluralist model, while the embrace of cost-benefit analysis sounds more in the key of civic republicanism. Regardless of the theoretical grounding, both have tended to promote the addition of new procedural embellishments to the existing notice-and-comment framework that produce suboptimal results. Specifically, they slow down the rulemaking process without improving the quality of regulatory decisionmaking, and they have reinforced power inequality by giving entrenched interests new tools for blocking or weakening new policies they oppose – that is, maintaining a status quo bias towards inaction. In turn, these consequences seem to be artifacts of a characteristic that pluralism and civic republicanism both share: an abiding belief that consensus can be achieved and should be the goal of regulatory decision-making.
Third Theory of Regulatory Democracy: Agonism
Yet, this shared belief has come under increasing criticism in recent years, leading scholars to entertain a third theory of regulatory democracy: agonism. This model begins from the premise that consensus is impossible to achieve in many policy contexts, particularly during times such as now marked by polarized discord and seemingly incommensurable division over values and worldviews. If conflict is inevitable, agonism posits, then the appropriate function for our democratic institutions is to channel that conflict so that it is as productive as possible. Under this view, the legitimacy of policy outcomes comes not from how they are ultimately resolved, since they will not be accepted as such by many stakeholders. Instead, legitimacy flows from affording dissatisfied stakeholders with an ongoing realistic opportunity to contest and displace those outcomes with those that more closely align with their preferences. Administrative law already contains agonistic features. Adherents of this model envision other institutional and legal reforms that would infuse a more agonistic orientation to regulatory democracy. For instance, they would require more frequent use of retrospective review for regulations and greater use of adjudication for policymaking in place of rulemaking where possible.
One of the practical benefits of redesigning regulatory democracy mechanisms consistent with agonism is that it could help prevent some of the abusive gamesmanship practices in the notice-and-comment process described above. By demanding consensus, prevailing theories of pluralism of civic republicanism create perverse incentives for entrenched interests to manipulate public participation mechanisms in order to prevent consensus from ever being achieved. For instance, such interests might seek to delay final action on a rule by packing the rulemaking record with the intent to manufacture uncertainty or continually raise new issues – rather than productively inform a regulatory decision. In this way, public participation becomes a tool for translating power into paralysis. This concern is well worth considering given, as adherents of Abundance liberalism have noted, such paralysis can be exploited by would-be authoritarians to support an agenda of democratic backsliding.
Modernizing Regulatory Democracy Through Public Participation Planning
In light of these challenges, it is time to consider not just incremental changes, but a fundamental rethink of how public engagement is conducted within the administrative state. In contrast to current approaches, effective regulatory democracy must be both people-centered and power-conscious.
This paper will concentrate on applying this reform framework to the rulemaking process, though it could also be applied to other aspects of the federal administrative state that involve public participation, such as NEPA and permitting. Likewise, they could be applied to state-level administrative analogs. While there might be several ways to build a people-centered, power-conscious, rulemaking process, this paper outlines what it refers to as Public Participation Planning. This would involve agencies:
- developing and executing a strategy that is
- tailored to each of their planned regulatory actions in order to
- engage relevant stakeholders throughout each step of the rulemaking process
- with the explicit purpose of building a reasonably comprehensive record of the public’s views on the action for the rulemaking record.
The validation of Public Participation Planning as a viable mechanism for achieving truly meaningful engagement comes through its embrace of four cross-cutting principles: proportionality transparency, communication, and pragmatic learning. First, the rigor of a particular plan should be fairly proportional to the significance of the rule under development. Second, strenuous adherence to transparency is essential for achieving the agonistic goal of productive disagreement. In particular, agencies must always be completely forthright with all stakeholders about how decisions were reached and what evidence and arguments proved determinative. Third, and related to transparency, agencies should strive to maintain open lines of communication with stakeholders throughout the entire rulemaking process. This will enable agencies to serve as the effective mediators of productive disagreement through the rule’s development and beyond.
Fourth, it requires agencies to commit to an ethic of pragmatic learning. Implementing Public Participation Planning is not as simple as plugging a few numbers into an equation and expecting an optimal result to emerge; it is impossible to predict ex ante what will work in any given situation. Instead, to make the most of Public Participation Planning, agencies will need to become adept at building and rebuilding the proverbial plane, even as they are flying it. Moreover, what has worked in the past will have to be continually reassessed in light of underlying power inequities. If history is any guide, powerful incumbents will eventually devise ways to use their resource advantage to corrupt even the best public participation mechanisms for engaging structurally marginalized communities.
As discussed in greater detail, one of the obvious objections to Public Participation Planning is that its apparent emphasis on proceduralism will exhaust scarce agency resources and contribute to excessive delays in a rulemaking process that has already become too bogged down to permit for effective and timely policy implementation. These four cross-cutting principles, however, are intended to alleviate those concerns – adherence to them will help to strike the needed balance between public engagement and expeditious policymaking.
Putting Public Participation Planning into Action
Public Engagement Strategy Blueprints
One of the distinguishing features of Public Participation Planning is the requirement that agencies assemble a public engagement strategy blueprint for engaging stakeholders for each planned regulation at the time that the action is initiated that is tailored to the unique circumstances of the rule. This step provides agencies with a mechanism to anticipate potential challenges and think through and identify effective solutions that are calculated to enable them to build a reasonably comprehensive record of the stakeholders’ views on the rule.
It is worth emphasizing at the outset that the development of a public engagement strategy blueprints need not be a resource-intensive and time-consuming task. As noted above, these should be tailored to match the significance and controversy level of the rule. In addition, agencies will learn by doing, resulting in increased efficiencies over time. For instance, analyses used for past rules will often be readily usable for future rules addressing similar subjects. Another crucial source of efficiencies will be for agencies to use their existing institutional resources to perform these tasks. Most rulemaking agencies already have various forms of public engagement offices and regional and local offices that can and should be tapped. Public affairs offices can also be brought into the rule development process earlier, rather than announcing decisions – such as proposals and final rules – after the fact. Lastly, agencies may build new institutions that increase efficiencies for implementing public engagement strategy blueprints. For instance, agencies may consider creating a standing process for conducting periodic townhalls or other listening sessions. This relatively modest investment could in turn yield significant value for informing the development of public engagement strategy blueprints for future rulemakings covering a wide variety of issues.
More broadly, as explained in greater detail below, the implementation of all aspects of Public Participation Planning, including the public engagement strategy blueprints, should be thought of in terms as an investment. There is no denying they will involve the dedication of resources and time – mostly at the front end of the rulemaking process. But, by directly addressing power disparities and by more constructively channeling irreconcilable disagreements over the competing values implicated by a given rulemaking, Public Participation Planning will mitigate the sources of delay that crop up later in the regulatory process, including, most notably, litigation and the problem of ossification it creates. And to be perfectly frank, much of this work involves things agencies should be doing anyway. The failure to do so often helps to explain why many agency rules have fallen short of accomplishing their stated goals. In short, more “process” at the beginning will lead to less process and less delay overall.
Step 1: Public Engagement Strategy Blueprint
The first step in assembling a public engagement strategy blueprint is to conduct a thorough and deliberate stakeholder mapping exercise. By the time an agency has completed this exercise, they should be in a position to identify the relevant range of stakeholders for a given rulemaking and the anticipated role or roles they might be expected to play in the rulemaking process. For each category of stakeholders, agencies should also perform a general capacity assessment. Specifically, they should seek to answer such questions as what kind of input or expertise members of a given stakeholder group are likely to bring, whether and how those individuals have participated in similar rulemaking processes in the past, and what barriers might prevent them from participating effectively in the current rulemaking process.
Step 2: Examine Power Disparities and Structural Injustice
The second step is to assess the role, if any, that underlying power disparities or other forms of structural injustice (e.g., racism or patriarchy) play in contributing to the problem that the regulation is meant to solve. To be sure, agencies should be performing such assessments anyway since a failure to do so could yield policy responses that either fail or have other unintended perverse effects, including making the underlying problem worse. With this background in place, the agency should then give careful consideration to how stakeholders identified through the mapping exercise might help them to better understand these underlying power disparities.
Step 3: Stakeholder Engagement to Inform Rulemaking
The third step is to use any learnings from the stakeholder mapping exercise and power disparities assessment to construct a strategy for conducting stakeholder engagement to inform the development of the rulemaking proposal before it is formally published. Most agencies already have an established “action development process” they follow for drafting proposals and building a supporting evidentiary record. Agencies can build off the procedural framework that process creates when designing this engagement strategy. They can ask which types of stakeholders are likely to have input that would help with the successful completion of each stage of the policy development process and what specific engagement tactics would likely be most successful in obtaining that input at a reasonable cost in terms of time and resources. For example, agencies might use more time-consuming and resource-intensive focus groups for particularly weighty matters such as scoping out alternative regulatory designs. In contrast, they might employ informal remote public hearings to quickly gather ideas for sourcing certain kinds of evidence related to the rulemaking. (A bonus of this process is that it might reveal ways in which an agency policy development process could be strengthened, by adding, removing, or combining steps, or by altering their order.)
In preparation for this step, agencies will likely also want to have created a general library or “menu” of engagement tactics, with a brief assessment of their strengths and weaknesses. This will enable agency staff to quickly pull tactics “off the shelf” and insert them into the individual public engagement strategy blueprint. Indeed, this is an example of how moving along the learning curve will help agencies to implement Public Participation Planning more quickly and at reduced cost.
The capacity assessment performed during the stakeholder mapping exercise will especially be important for successfully implementing this step of plan development. For instance, if that exercise revealed that an important group of stakeholders is unlikely to have reliable access to high-speed internet, then the agency should refrain from relying on something like a remote public hearing to obtain input from those stakeholders. This assessment will also help agencies to identify potential affirmative steps they can take to eliminate barriers to public participation. For example, agencies can take steps to provide translation services if a large number of crucial stakeholders do not speak English as a first language. At the same time, the capacity assessment might reveal that a particular stakeholder group exercises an unusually high degree of dominance over a particular issue. In such cases, the agency may find that imposing certain constraints on their participation during the pre-proposal time period. These might include limiting or barring ex parte contacts or placing reasonable page limits on documentary submissions. (Such actions will also have the advantage of expediting the rulemaking process by preventing well-resourced contacts abuse these contacts as a means for delay.)
Step 4: Identify Mechanisms to Include Marginalized Communities, Including Storytelling
The fourth step in building a public engagement strategy blueprint is to identify mechanisms for ensuring that even stakeholders from structurally marginalized communities are able to participate in the notice-and-comment process as effectively as possible. As noted above, the notice-and-comment procedures often systematically exclude such individuals. While agencies cannot completely obviate this dynamic, they should still strive to sand off its worst effects – especially as these procedures are likely to remain part of the rulemaking process for the foreseeable future. Somewhat regrettably, the general consequence of these auxiliary mechanisms would be to get ordinary individuals to more effectively behave like sophisticated lobbyists instead of their true, authentic selves. This means providing various kinds of educational resources and specialized training to individuals so that their input can fit the “technocratic” mold, much as the Federal Energy Regulatory Commission’s (FERC) Office of Public Participation (OPP) undertakes now. It might also involve creating institutional mechanisms to serve as representatives or ombudsmen on behalf of unaffiliated individuals, though this would likely require significant additional resources and perhaps even legislative change to effectuate.
A more radical option would be to undertake institutional reforms that make notice-and-comment procedures more amenable to obtaining and utilizing non-technocratic forms of input, such as storytelling. This approach has the advantage of permitting individuals to share their more authentic expertise – including their situated knowledge and lived experiences – though such input may be of limited relevance at this later stage in the rulemaking process. The degree of institutional reforms required to fully realize this procedure – ranging from changes in agency hiring practices to modifications of administrative law doctrines to recognize these different kinds of “expertise” – makes it unlikely that this approach will bear fruit any time soon.
Step 5: A Plan for Public Engagement After Rulemaking
The fifth and final step in building a public engagement strategy blueprint is to create a plan for how the public might remain engaged after the rule is finalized – that is, to identify opportunities, if relevant and possible, for the public to participate in the rule’s implementation and ensure those are reflected in the rule’s final design. Examples of such engagement include the public’s role in monitoring compliance, measuring the rule’s impacts through citizen science activities, and holding regulated entities accountable for violations of the rule’s requirements through citizen suits when legally available. The final rule may also seek to explicitly incorporate opportunities for the public to participate in any future retrospective review actions for the rule, though Congress will need to ensure agencies receive sufficient budgetary resources to carry out such reviews. Similarly, many statutes authorize agencies to grant individual businesses different kinds of compliance relief, such as deadline extensions, variances, waivers, and exceptions. The final rule could provide the public with a meaningful role in considering and awarding these grants of relief.
As noted above, the rigor and detail of the blueprint should be roughly proportional to the rules’ economic and social consequences as well as to the level of controversy it is anticipated to engender. As with other aspects of implementing Public Participation Planning, accomplishing this proportionality goal in practice will improve with practical experience.
Resource constraints and political pressure for expeditious policy implementation are likely to provide strong incentives for agencies to fall short of the desirable level of rigor and detail. Consequently, countervailing incentive structures will be necessary to offset that tendency. Perhaps that could be accomplished through well designed judicial review standards, as noted below. Political leadership – including from the White House agency appointees – could also signal the importance of careful implementation of Public Participation Planning. For instance, this could be institutionalized through agency strategic planning exercises or encouraged as part of performance review and promotion decisions for career staff. Of course, Congress can do its part by fully funding agency implementation. And over time, as agencies advance along the learning curve for implementation, they will achieve increased efficiencies that will alleviate some of the incentives to do insufficiently rigorous Public Participation Planning.
Strategy Blueprint Implementation, Tracking, and Public Participation Plan Statements
As indicated above, each public engagement strategy blueprint that an agency develops should focus on creating meaningful participation opportunities for members of structurally marginalized communities early in the pre-proposal process, since that is when their input is likely to be of greatest relevance and utility for agency decision-makers. Rather than be a mere “check the box” exercise, the execution of these early participation mechanisms (informal hearings, focus groups, etc.) should have a discernible impact on the structure and substance of the proposal. Thus, as agencies turn to implementation of these mechanisms, they should carefully track whether and to what extent the public engagement strategy blueprint is accomplishing what they expected it would.
This, of course, is not to say that the agency should use these engagement activities to build evidence for decisions that were already made by other means – much as occurs with cost-benefit analysis now. Rather, it means that agencies should base their monitoring on other more objective benchmarks. One question agencies should ask is whether the quantity of participants matches the predicted expectations. (Again, agencies will likely struggle at first to make these kinds of predictions with much accuracy – there will be a learning curve. But, as noted above, a crucial ethic for Public Participation Planning is a commitment to learning by doing.) Similarly, agencies should find that the input they are receiving through these early engagement mechanisms are providing answers to the questions they need to answer to develop the proposal – whatever those answers happen to be. Another good indicator that the early engagement mechanisms are working well is that they are uncovering important “unknown unknowns” – things that the agency did not realize it did not know when it launched the rulemaking.
If, on the other hand, agencies are not finding that the early engagement mechanisms are working as expected – that they are not helping to build a reasonably complete record of public input on important policy-relevant questions undergirding the proposed rule – then they should make adjustments to the engagement strategy. This goal, of course, does not mean agencies should strive to accomplish something akin to comprehensive accounting for all relevant views from the impacted public. Instead, the goal should be to obtain a reasonably representative level of input from each of the major stakeholders included in the agency’s initial mapping exercise. What constitutes a reasonable level of input will necessarily be a subjective determination, and one that agencies will improve on as they learn through implementation of the Public Participation Planning scheme over time. In making this determination, though, agencies will want to be especially attentive to the concern that they have not adequately engaged members of stakeholder groups they have initially identified as being structurally marginalized or as facing particularly high barriers to participation. When in doubt, an agency may wish to attempt other forms of engagement for these groups. As in other forms of research, if the input they obtain sounds repetitive, that would indicate a good stopping point has been reached.
Drawing on lessons learned from actual practice, agencies may want to consider employing different forms of affirmative outreach to targeted stakeholder groups, undertaking alternative engagement tactics, or finding other creative ways to minimize barriers that might be preventing effective engagement. For example, if an important stakeholder category is young families, then the agency may consider securing resources to provide childcare during in-person hearings. To be sure, agencies may encounter legal constraints that may prevent them from instituting strategies like this. One suspects that these constraints are not as significant as feared, however, and that agency counsel have been overly cautious in interpreting these constraints. Nevertheless, clarifying legal authority from Congress on these matters would be welcome.
As it carries out the specific components of its public engagement strategy blueprint, the agency should begin assembling a comprehensive Initial Public Participation Plan Statement, which documents its outreach and engagement activities, carefully summarizes the input that was received through each component, and briefly explains what impact, if any, that input had on the agency’s proposal. Consistent with the principles of transparency and communication noted above, it is particularly important that the agency use this document to identify instances when a stakeholders’ input did not influence a particular outcome and explain why that was the case.
Explaining the Democratic Basis for a Rule
The agency should include the completed Initial Public Participation Plan Statement in the rulemaking docket when the rule proposal is formally published so that it is available to the public when they are developing the comments. In this way, the Initial Public Participation Plan Statement will function similarly to an Initial Regulatory Impact Analysis (i.e., the initial cost-benefit analysis), only it explains the “democratic” basis for the rule instead of its “economic” basis. Ideally, as the Initial Public Participation Plan Statement becomes more institutionalized, it can even replace the Initial Regulatory Impact Analysis that agencies now perform as the most prominent supporting document for a proposal. This would conserve agency resources and symbolize that democracy has replaced technocracy as the key driver of regulatory decision-making.
After the proposal is published, agencies should likewise carefully monitor the implementation of any specific components from the public engagement strategy blueprint for supporting public participation while the public comment period is open. Again, they should strive to make appropriate adjustments whenever they discover that these mechanisms are not producing expected or helpful results. During this period, agencies should continue documenting their progress in implementing the public engagement strategy blueprint by updating Initial Public Participation Plan Statement.
In conjunction with releasing the final rule, agencies should then include in the rulemaking docket a Final Public Participation Plan Statement. (Again, this final statement would be democratic analog to the Final Regulatory Impact Analysis.) This document should describe the public engagement strategy blueprint that was originally created, any changes that were made during the rulemaking process, what input was received through the agencies’ engagement mechanisms, and what impact they had on the proposed and final rules, if any. Again, agencies should be forthright in identifying the input that did not impact the rule and briefly explaining why.
Lastly, after the final rule has been published, agencies should dedicate resources and time to reflecting on lessons learned from the implementation of public engagement strategy blueprint. They should be prepared to incorporate these lessons into the design and implementation of future public engagement strategy blueprints. This will lead to implementation of Public Participation Planning that is more effective, less expensive, and quicker. In addition, agencies will also need to be prepared to track the implementation of any public participation mechanisms related to implementation incorporated into the final rule design. As noted above, these mechanisms might relate to compliance monitoring and enforcement, retrospective review, and grants of compliance relief.
Advantages of Public Participation Planning
Public Participation Planning stands in stark contrast to the largely one-sized-fits-all approach to public engagement – basic notice-and-comment procedures with occasional public hearings – that characterize the current rulemaking process. As noted above, essentially no deliberation goes into the creation of this engagement strategy – it is effectively reflexive – nor does it recognize, much less attempt to address, realistic concerns that important categories of stakeholders may not be accounted for in its strategy or that such incomplete input risks aggravating the very power disparities and social inequities that gave rise to the problem that the rule is meant to address in the first place.
In addition, successful implementation of Public Participation Planning will promote better regulatory democracy in the following ways. First, it will provide agencies with a mechanism for systematically identifying all the relevant stakeholders for a given policy, particularly members of communities who might otherwise be systematically excluded from such decision-making processes by structural or other barriers. Second, it will ensure that input is elicited from these stakeholders consistent with the sequential logic of the rulemaking process, providing agency decision-makers with the information they need when it is most useful.
Third, it will empower agencies to tailor their outreach and engagement strategies to the unique policymaking context implicated by the rule under development. Fourth, it will enable agencies to use public engagement to surface and account for any underlying power disparities that contribute to the policy problems a rule is meant to address, leading to more effective and durable policies. Fifth, it will highlight productive disagreement among stakeholders rather than engage in a quixotic pursuit of consensus – that is, it seeks to move regulatory democracy in a more agonistic direction. This is essential to recalibrate public engagement so that it is more attentive to power disparities and to avoid being a source of excessive delay in the policy development process.
How Other Federal Institutions Can Support the Successful Implementation of Public Participation Planning
The White House
With the advent of presidential administration under Reagan, the White House has played an increasingly active role in coordinating and steering the actions of the administrative state. The White House would thus be well-positioned to support the effective implementation of public participation planning. Indeed, as noted above, the Biden administration took some important initial steps on strengthening public participation in the rulemaking process as part of its broader Modernizing Regulatory Review initiative.
A logical place to start would be for staff at the White House Office of Management and Budget (OMB) to produce a comprehensive list of public outreach and engagement tactics for agencies to use to inform their own public engagement strategy blueprints. They could create this list by surveying the relevant academic literature, reviewing agencies’ past experiments with innovative approaches, and even looking at examples offered by peer democratic states abroad.
To support ongoing agency learning, OMB could also convene a standing working group composed of representatives from the public engagement offices at the various agencies. This working group could provide a forum in which these offices regularly share their best practices and lessons learned. Just as significantly, by signaling that public engagement is a priority of administration leadership, the working group would also by its mere presence help to reinforce a broader ethic and commitment to democratic inclusiveness across the administrative state.
Inviting OMB support in the implementation of Public Participation Planning is not without risk, given its historic role of interfering with and unduly politicizing the rulemaking process. It would certainly be preferable if Congress created a new standalone office outside of the White House that is explicitly charged with these tasks, as suggested below. But short of that, OMB is institutionally best positioned to play this role – provided that it does so in a strictly auxiliary fashion, leaving individual agencies the ultimate discretion on how to implement Public Participation Planning. In addition, carrying out such an auxiliary role would be a far better use of OMB’s resources than its current practice of superintending agency decisionmaking through the centralized regulatory review process.
The implementation of Public Participation Planning would also benefit greatly from having staff with different kinds of skillsets and life experiences. For instance, staff with backgrounds in social work or community organizing and specialized training in sociology might be particularly valuable. The White House Office of Personnel Management (OPM), the main human resources agency for the administrative state, could be instrumental in helping agencies to identify and hire such individuals. OPM could also help make necessary revisions to hiring standards and practices to make it easier and quicker to bring them on board.
Congress
Agencies have adequate legal authority to undertake Public Participation Planning. Still, Congress can ensure that even future administrations that might be hostile to the goals of regulatory democracy will implement this reform, even if reluctantly, by codifying this procedure into law through an amendment to the APA.
Similarly, implementation of Public Participation Planning likely would not require a significant commitment of agency resources – especially, if agencies are able to redirect resources to it from other rulemaking requirements, such as cost-benefit analysis or the myriad energy-related analyses that agencies must conduct pursuant to various executive orders. Ideally, Public Participation Planning will also reduce the incidence of legal challenges against final rules, which would promise to save on direct litigation costs. With these reduced litigation risks, agencies may also find that they are no longer compelled to “bulletproof” their rules through elaborate rulemaking records and gargantuan preambles to their final rules. This resulting streamlining effect of Public Participation Planning could also yield significant cost savings for agencies over the long run.
Nevertheless, Congress should still commit adequate appropriations for agencies to launch this reform, especially while they are still overcoming the incremental additional costs required to move through the early stages of the learning curve. With increased experience and specialization, agencies will likely be able to implement Public Participation Planning in an increasingly cost-effective manner.
Congress can take other steps to affirmatively support Public Participation Planning. For instance, they can authorize and fully fund a new institution that affirmatively supports public participation in the notice-and-comment process. The EXPERTS Act, a comprehensive progressive regulatory reform bill now pending in Congress, offers one potential model. Specifically, it would create something called the Office of the Public Advocate, which would be charged with this responsibility.
In addition, Congress can tap the Administrative Conference of the United States (ACUS) – which is effectively the federal government’s in-house “think tank” on administrative law – to study existing administrative law doctrines that might present a barrier to effective implementation of Public Participation Planning (to the extent that the doctrines arise from statutory, as opposed to constitutional, law). Such doctrines might include Vermont Yankee’s bar on judicially created administrative procedures (the codification of public participation planning, recommended above, would obviously address this), Loper Bright (which gives the judiciary, instead of agencies, the primary responsibility in interpreting agencies’ statutory authority), and the “logical outgrowth” test (which constrains how significantly a final rule’s substance can deviate from what’s contained in the proposal). ACUS could develop recommendations for how agencies and reviewing courts can avoid running afoul of these doctrines or propose legislative fixes for Congress to adopt.
Lastly, it goes without saying that Public Participation Planning would benefit immeasurably from having a functional Congress in place. According to the common conservative myth, an empowered Congress is necessary to restrain the administrative state. Just the opposite is true, however. By recommitting to doing the people’s business and passing public interest legislation again, Congress would provide agencies with fresh opportunities to put Public Participation Planning into action with up-to-date legal authority to tackle the new and emergent problems that pose a great threat of harm to structurally marginalized populations.
The Judiciary
The most obvious way that the judiciary could support the implementation of Public Participation Planning is by devising new judicial review doctrines that reward rulemakings with exceptional democratic pedigrees with enhanced levels of deference. This, of course, would require conservative judges to apply an even hand to all regulations challenged on judicial review before them. That means they would have to deploy enhanced deference consistently and in the service of promoting regulatory democracy, rather than wield it as a weapon to justify striking down rules they oppose on policy grounds. Under present circumstances, one might be forgiven for doubting this will take place. Yet, since we cannot avoid this institution either, we must still do the best we can with our politicized judiciary until such time as we are able to accomplish significant judicial reform – a topic beyond the scope of this paper.
Such enhanced deference might have a role to play in assessing the statutory authority for the agency’s rule, even under the new Loper Bright review framework. For instance, reviewing courts might modify the application of Skidmore to apply a special degree of “respect” for agency interpretations that rest on public input received during the rulemaking process.
More likely, though, the influential weight of public input would be greatest during the review of agency policy decisions under the arbitrary-and-capricious standard. In theory, courts already employ a “super deference” for agencies’ determinations based upon science and other forms of technocratic expertise, though courts rarely follow this approach in practice. Courts could easily create (and actually follow) an analogous super-deference doctrine for agency determinations based on “democratic expertise.” The inclusion of the Final Public Participation Plan Statements in the rulemaking record, as outlined above, would provide the essential informational foundation for the application of such a doctrine. In developing this doctrine, courts would have to account for applicable doctrinal constraints, including, most notably, Vermont Yankee.
Short of that, though, much of the analysis involved in assessing Final Public Participation Plan Statements would fit comfortably within the “hard look review” that courts already perform as part of the arbitrary-and-capricious standard of review. In other words, implementation of Public Participation Planning would not create any insurmountable barriers for courts conducting judicial review pursuant to the APA.
Courts have long recognized that the APA’s arbitrary-and-capricious judicial review standard implies a general duty for agencies to build a sufficiently complete rulemaking record to enable such review. Contained within this broader duty is a more specific responsibility to have procedures or mechanisms in place for ensuring that the information before the agency meets some minimal level of quality. While this responsibility might traditionally be thought of as applying to more technocratic inputs, there is no reason why it should apply equally to the unique on-the-ground expertise of individuals and community-based organizations. Similarly, this general duty also implies a more specific requirement that agencies ensure that the scope of information available to them be sufficiently broad to permit for evidence-based, reasoned decision-making required by arbitrary-and-capricious review. Again, this concern should apply equally to all forms of expertise, not just those regarded as technical or scientific in nature.
Importantly, this same judicial review standard would also guard against attempts by any president who is hostile to regulatory democracy to implement Public Participation Planning with insufficient rigor. Just as the Trump administration is now seeking to bypass the notice-and-comment process altogether, a future administration might reduce this process to a mere check-the-box exercise or conduct woefully inadequate outreach and consideration of input. The Final Public Participation Plan Statements would afford a reviewing court with a basis for applying the arbitrary-and-capricious standard to the agency’s public engagement efforts and, ultimately, to remand an agency rulemaking to correct this aspect of the record where any flaws or gaps are identified.
The notion of courts policing Public Participation Planning raises a separate concern that this aspect of arbitrary-and-capricious review could be abused by conservative activist judges who are opposed to climate policy or other aspects of the progressive policy agenda. The Supreme Court’s recent extreme application of the arbitrary-and-capricious standard in Ohio v. EPA confirms that this is not an idle concern. Still, it seems clear that activist judges will find plenty of opportunities for abusing arbitrary-and-capricious review even in the absence of Public Participation Planning. On balance, then, the benefits of this reform would still seem to outweigh these risks.
Public Participation Planning as Part of a Broader Agenda to Increase Administrative Effectiveness
The past year has seen the Abundance Liberal movement spark a robust debate within the broader liberal community over the appropriate role of legal procedure in our governing institutions. Under the circumstances, then, it may seem like an unusually inopportune time to champion something like Public Participation Planning – an essentially proceduralist reform. As explained below, though, this reform strives to take seriously Abundance’s critiques and is consciously predicated on the recognized imperative to strike an appropriate balance between, on the one hand, public engagement and, on the other, effective, responsive administrative action that delivers concrete results.
The Abundance Liberal movement, as best captured in the recent book by its most prominent advocates Ezra Klein and Derek Thompson, argues that the Left’s reflexive embrace of proceduralism and litigation is an antiquated relic from a bygone era and is already becoming a political liability. That is because many of the progressive movement’s policy priorities – from addressing the climate crisis to promoting affordable housing – requires quick policy implementation, a goal that is ultimately defeated by excessive proceduralism. Instead, the book argues, the Left should dispense with most procedures as a mechanism for legitimizing government action and instead let the popular results of those actions (e.g., affordable housing, cheap clean energy, etc.) serve that legitimizing function after the fact.
Significantly, Klein and Thompson’s book singles out public participation in the policymaking process as emblematic of the broader problem they are trying to solve. Indeed, their specific critiques of notice-and-comment procedures largely tracks with those that have motivated the proposal Public Participation Planning, as detailed above. In particular, they correctly identify these procedures as excluding structurally marginalized communities and reinforcing broader power disparities in our society.
Where the Public Participation Planning proposal departs from Abundance adherents such as Klein and Thompson is its core claim that the problem is not procedure per se, but power disparities. More specifically, it posits that the dysfunctional procedures that cause excessive and unnecessary delays in policy implementation are better understood as a symptom of the deeper problem of power disparities in our society. After all, even if we were to remove all existing procedural requirements – that is, to take Abundance to its logical extreme – it is by no means clear that we would see more expeditious policy implementation, particularly where those policies go against the preferences of entrenched elites. Such interests would simply find other, non-procedural mechanisms for blocking policies they opposed.
Given that we are unlikely to eliminate the structural sources of power disparities in our society any time soon, it is worth exploring more practicable near-term mechanisms for alleviating the worst consequences of those power disparities. What Abundance potentially misses with its black-and-white diagnosis of the procedure problem is that procedure actually holds a lot of promise for accomplishing this goal. After all, if procedures can aggravate power disparities, as Abundance Liberals would freely stipulate, then it also follows that well-designed procedures can do the opposite as well. Legal scholar Nicholas Bagley, who has provided part of the intellectual foundation for Abundance, highlighted this intrinsic feature of procedure when he wrote: “government action — whether it involves dispensing public benefits or regulating private conduct — allocates resources, risk, and power within the United States.”
To put Bagley’s point differently, procedure can never be neutral in its effects on underlying power dynamics; it will tend to cut in the favor of one set of stakeholders or another. Taking this reality seriously means that policymakers have a lot of tools at their disposal to shape and reshape those power dynamics in more productive ways through carefully designed procedures. More to the point, Abundance overlooks the tantalizing possibility that, by effectively redistributing power within the administrative state, well-designed procedures can actually expedite policy implementation – or at least add value, such as improving the quality of decision-making, without causing new or undue delay. This is precisely the project that Public Participation Planning sets out.
As described above, Public Participation Planning illustrates how procedures might be designed with the goal of affirmatively redistributing power from entrenched interests committed to maintaining a suboptimal, unjust status quo to members of structurally marginalized communities that are both underrepresented in our political processes and disproportionately burdened by the harms that arise from status quo economic, social, and political arrangements. Specifically, it seeks to carve out new spaces in the rulemaking process for bringing in the unique expertise of historically underrepresented populations at precisely the points in that process when their expertise is most germane. At the same time, it contemplates placing reasonable constraints on the participation opportunities available to entrenched powers such that their input is channeled to maximize its utility for agency decision-making. This would also have the additional benefit of preventing these interests from abusing their resource advantages to overwhelm agency decision-makers with extraneous information for the purpose of causing unnecessary delay.
Public Participation Planning’s commitment to transparency and ongoing communication with stakeholders plays an important contributing role in this power shifting dynamic. For structurally marginalized stakeholders, it will be essential for them to know concretely how their input is materially shaping the policy and factual determinations that undergird agency decision-makers. Otherwise, it will be entirely rational for members of these communities to assume that their participation is little more than a check-the-box exercise, rather than a genuine effort at promoting regulatory democracy. Understandably, members of such communities will still harbor some degree of skepticism even despite agencies’ good faith commitment to transparency and communication. Trust will take time to build, but it can be built. Somewhat counterintuitively, one way that agencies can do this is by always honestly explaining to these stakeholders when their input did not substantively influence a particular decision – that is, by delivering the bad news as well as the good.
Equally as important, Public Participation Planning demonstrates how these kinds of investments in well-designed procedures can actually pay off in terms of more expeditious policy implementation. In other words, it helps to refute the commonly held belief – one clearly embraced by the Abundance Liberal movement – that public engagement and expeditious policy action are fundamentally at odds. Instead, these tradeoffs can effectively be mitigated when such public engagement procedures are designed to correct power disparities that are implicated by a given rulemaking.
The key to accomplishing this seeming procedural alchemy is by addressing the primary driver of rulemaking delay: litigation over final rules. (To be sure, Public Participation Planning would also endorse Abundance’s more general call to clear out unnecessary procedural requirements that have accreted over the years, such as cost-benefit analysis, the Regulatory Flexibility Act, and the Unfunded Mandates Reform Act, to name a few. Doing so would be entirely consistent with its analytical framework. These procedures tend to aggravate power disparities – often by design – and thus contribute to delays in the rulemaking process.) Litigation is time-consuming in its own right, often taking several years to reach a definitive conclusion. During this time, agencies are increasingly subject to court orders barring them from implementing the rule’s provisions.
In addition, as noted above, the near certainty of legal challenges creates strong incentives for agencies to “bulletproof” their rules to reduce the chances they will be struck down on judicial review. While some degree of deliberative and evidence-based rigor underlying rules is desirable, of course, the perverse result of this incentive structure is that agencies go far beyond what the law reasonably requires for substantiating rules, an increasingly resource-intensive undertaking that significantly delays the completion of new actions.
As noted above, many of Public Participation Planning’s distinguishing features are designed to reduce the incidence of litigation. Particularly significant in this regard is its emphasis on early engagement. Not only does engagement provide agencies with better and more timely input; it also has the additional benefit of promoting greater buy-in from stakeholders, which in turn defuses litigation risk down the road. In other words, frontloading public engagement seems to shorten the length of the rulemaking process even if agencies are technically conducting a greater number of procedural steps overall.
Several empirical studies of the implementation of the National Environmental Policy Act’s (NEPA) analytical requirements appear to confirm this effect, finding that NEPA processes that involve early public engagement are statistically shorter than those that do not. Reduced litigation and, by extension, the reduced incentives for bulletproofing NEPA analyses seem to explain this discrepancy. (Not incidentally, extensive NEPA-related delays are also the subject of extensive criticism by the Abundance movement) It is reasonable to expect that this dynamic would translate to the functionally similar rulemaking context as well.
Also important, Public Participation Planning seeks to institutionalize a more agonistic orientation into the regulatory process. As explained above, agonism seeks to avoid the pursuit of near-universal consensus over policy outcomes, which is typically impossible to achieve in practice anyway, and instead create conditions for productive disagreement. Similar to early engagement, one of the desired effects of administrative agonism is to reduce litigation risk and the perverse effects such risks create. Instead, final regulations would be treated as more contingent and subject to realistic mechanisms for ongoing revision and refinement or to various forms of implementation flexibility. In this way, agonism enabled by Public Participation Planning would attempt to lower the stakes on final rules, such that stakeholder focus could be gradually shifted from post-finalization litigation to implementation where it can be put to more productive use.
To be sure, achieving the full agonistic potential of Public Participation Planning would require other legislative changes. For instance, Congress could amend agencies’ authorizing statutes to give them greater authority to deploy back-end implementation adjustments and flexibilities, such as waivers, exemptions, and compliance extension deadlines (all subject to vigorous public participation mechanisms, as noted above). For agencies that already enjoy these kinds of authorities, implementing this general approach could serve as a valuable proof-of-concept demonstration that could help catalyze this legislative action in the near future.
Another option would be for Congress to incorporate more rigorous schedules for reviewing and updating regulations into their statutory design, such as those that exist for appliance energy efficiency standards. To ensure that these reviews are carried out expeditiously, Congress could also experiment with different kinds of “hammer provisions” that would kick in automatically – setting default regulations or standards, for instance – if an agency is unable to work with relevant stakeholders to adopt the update according to the statutory schedule. Ultimately, the goal of these reforms would be to rebuild a rulemaking process in which stakeholder contestation is increasingly shifted to the implementation phase, where dynamism and flexibility can be permitted to flourish.
Even better, the judiciary could further reinforce this litigation-dampening effect of Public Participation Planning by adopting a strong deference doctrine based upon a rule’s democratic pedigree, as noted above. Such doctrines have the potential to substantially alter the calculus for stakeholders who are contemplating a challenge against the rule. If a rule’s legal and policy bases are strongly supported by public input such that it is likely to earn some measure of enhanced judicial deference, stakeholders may refrain from undertaking the expense of bringing a legal challenge, given the reduced likelihood that the challenge would succeed.
Even with all this careful attention to the design and implementation of Public Participation Planning, ongoing vigilance from policymakers will still be required to ensure that its performance delivers on its promises of better-informed decision-making and more expeditious policymaking. Regrettably, while effective use of this tool can help to alleviate the consequences of power disparities, those underlying power disparities will still remain in place, absent other more radical power interventions. The practical upshot is that over time entrenched interests may learn to “capture” parts of the Public Participation Planning program and deploy them to advance their narrow policy preferences at the expense of the broader public. (Both the APA notice-and-comment process and NEPA’s analytical requirements appear to illustrate this general dynamic.) This is why Public Participation Planning demands that agencies continually reevaluate and update their public engagement and outreach actions. Ideally, though, the general Public Participation Planning framework will remain resistant to such capture risks, even if more specific tactics and strategies for instituting that framework do not.
Conclusion
An effective response to the climate crisis faces numerous obstacles. One of these is a regulatory system that inadvertently reinforces underlying power disparities that help to maintain status quo conditions on how we obtain and use energy. A better approach to the practice of regulatory democracy – one that takes seriously and affirmatively addresses power disparities – will be essential for overcoming this obstacle.
This paper proposes a comprehensive reform to how agencies engage the public during the rulemaking process called Public Participation Planning. The distinguishing feature of this reform program is that it would require agencies to develop tailored public engagement strategy blueprints for each of their planned rulemakings. The purpose of these blueprints is to enable agencies to draw on a variety of engagement tactics that are calculated to build a reasonably comprehensive record of the stakeholders’ views on the rule. Importantly, the blueprint would also enable agencies to determine when best to engage different stakeholders throughout the various stages of the rulemaking process – a concept known as sequential participation. This reform would also require agencies to assemble Initial and Final Public Participation Planning Statements, which document the implementation of the blueprints and the ultimate impact that the resulting public input had on the substance of the rule. These Statements would be made part of the rulemaking record where they could be considered by judges, if necessary, during judicial review of the rule.
Public Participation Planning offers two major advantages. First, and most directly, by improving the quality of public input, it will lead to better decision-making, and thus better policy outcomes. Second, its implementation is likely to expedite rulemakings in many instances. Public Participation Planning would help to accomplish this outcome by using procedures to alleviate power disparities among relevant stakeholders. Properly understood, such power disparities appear to be the root cause of delayed rulemakings in the past. Taking these two advantages together, Public Participation Planning would offer a crucial piece in the larger puzzle of addressing the climate crisis effectively and with the urgency needed to avoid its worst consequences.
CELS Playbook: Clean Electricity for Local and State Governments
State and Local Actions to Make Government Work for People, Reduce Utility Bills, and Deploy Clean Energy
Elected leaders across the country are staring down interlocking crises. Families and businesses are struggling to pay skyrocketing utility bills. Large new demands are straining the grid and overtaking the buildout of new power plants. And the public’s faith in government has hit new lows. We need a new playbook to solve these problems and make the government responsive to peoples’ needs.
What’s going wrong?
Utility bills are rising rapidly for households and businesses due to an administrative state ill-equipped to protect customers from costs and risks. The cost of power supply is increasing due to growing demand, long timelines to build new cheap clean energy, and volatile natural gas prices. Utilities are spending more money on the transmission and distribution grid for both maintenance and recovery from wildfires and other disasters. Today’s regulatory construct allows utilities to drive spending decisions and pass on all these costs to customers, and regulators are under-resourced and unwilling to find alternative solutions.
Meanwhile, we are not building clean energy nor upgrading the grid fast enough to meet demand growth and address climate change. And this problem will get worse as power-hungry data centers connect to the grid and electrification of buildings, vehicles, and factories adds additional electricity demand.
The old climate policy playbook is not equipped for this moment. While it has driven significant deployment of low-cost clean energy, it was not designed to address non-financial obstacles to building projects and upgrading the grid nor to fully mobilize the suite of finance tools needed for the energy transition, nor to demonstrate that the government can make peoples’ lives better, now and long term.
Where do we go from here?
Policymakers and advocates need an expanded playbook. One that addresses the full set of barriers impeding financing and construction of clean energy and grid modernization projects. One that targets the root causes of high energy costs. One that reworks the administrative state to make government work for the people.
FAS, with the help of partner organizations spanning ideology and function, launched the Center for Regulatory to Ingenuity to build a vision for a government that is agile and responsive and delivers affordable energy, abundant housing, and safe transportation for all Americans.
As part of this work, we have developed an updated set of policies and actions for state and local leaders to meet this moment. We started by identifying the barriers to deployment and the flaws in the old playbook, published in our report Barriers to Building. Now we are developing the “plays” in a new playbook—tangible actions that state and local leaders can take now to make near-term progress and pilot new solutions. These plays will live on this landing page, which we will continue to update with additional actions.
This playbook is not a laundry list of policies but rather a cohesive strategy to achieve two goals: (1) deploy the clean energy and grid upgrades necessary to make energy affordable and combat climate change and (2) create governments that tangibly improve peoples’ lives.1
Contents (click to jump to a section)
- Main Character Energy: Make Regulators Main Characters in Planning and Ratemaking
- Improve State Government Responsiveness to Clean Energy Projects
- Build Administrative Capacity to Plan for an Affordable & Reliable Grid
- Fix Broken Incentives to Expand Distributed Energy Resources
- Wield Creative Finance Tools to Drive Investment and Reduce Capital Costs
Main Character Energy: Make Regulators Main Characters in Planning and Ratemaking
Utilities and their regulators are responsible for major decisions about what infrastructure we build and how much people pay for energy. Utilities—which can be owned by investors, the public (e.g., municipal utilities), or members (i.e., electric cooperatives)—conduct detailed analysis and provide proposals on planning and ratemaking to their regulators. The set of solutions below focuses on investor-owned utilities, who are incentivized to prioritize projects that maximize the returns for their shareholders. As a result, they underutilize solutions that could save customers money but do not earn companies a profit, like rooftop solar or technology-or maintenance-based upgrades to existing transmission lines.
In a well-functioning system, regulators—whether Public Utility Commissions (PUCs) or locally elected officials—would rigorously interrogate utility analyses and direct the utilities to shape or revise their proposals to maximize benefits to the public at lowest public cost. This lens is needed to ensure that utilities are spending money wisely in the public interest and prevent unnecessary bill increases from overspending on the wrong solutions. Active regulators are also needed to incorporate long-term considerations in planning and ensure consideration of strategies that provide long-term benefits. However, regulators are often not well-equipped or politically willing to conduct detailed analysis and push back on utility proposals. Other intervenors, like consumer advocates and environmental organizations, are outspent by the utilities, who can recover the costs of their analysis and interventions through customer bills in most states.
The result is a reactive, short-term-focused administrative state that leaves the public frustrated. Regular people are frustrated with skyrocketing bills, clean energy companies are frustrated with slow processes and broken incentives, and both are frustrated with the government’s ability to solve big problems. The administrative state itself—the officials and staff who make up regulatory body and state and local governments—are also frustrated with their perpetually reactive role and with limited say in outcomes.
We should not accept the status-quo regulatory process as a given. As representatives of the public, regulators should have both the ability and motivation to actively drive toward an abundance of cheap clean energy, affordable bills, and a modernized reliable grid. Achieving this vision requires the right personnel, clear direction and support from governors, and adequate analytical capacity.
Solutions
I. Direct Regulators to Use All Tools to Lower Energy Bills and Deploy Clean Energy (Governors and Legislatures)
When regulators take a backseat and let utilities drive, they narrow the toolkit of resources that can help meet demand and as a result leave savings on the table. Regulators with a mandate to prioritize affordability and clean energy buildout can reduce bills by both better scrutinizing utility plans and taking a more active role in enabling clean energy deployment. This includes finding creative tools to get more out of the grid through distributed energy resources, alternative transmission technologies, and flexible sources of demand like electric vehicle charging and factories with electric appliances.
Governors can direct PUCs to audit utility investments to find opportunities for savings, re-evaluate utility business models and incentive structures, consider distributed energy resources and alternative transmission technologies in planning, and consider climate impacts in planning and ratemaking decisions.
Legislatures can set statutory requirements for PUCs to consider these opportunities and expand their mandates to include clean energy goals and highest net benefit criteria.
Legislators can require PUCs to find savings across the gas and electric systems, including by using beneficial electrification to reduce costs.
In 2021, the Maine legislature directed the PUC to consider emissions reduction targets and equity impacts in regulatory decisions.
Oregon Governor Brown directed its PUC to integrate the state’s climate pollution reduction goals and promote equity by prioritizing vulnerable populations and affected communities.
New Jersey Governor Sherrill directed the PUC to review utility business models and assess whether they are aligned with cost reductions for customers.
In 2024, Minnesota S.F.4942 mandated that the PUC establish standards for sharing utility costs for system upgrades, ensuring fair cost-sharing and advancing state renewable and carbonfree energy goals along with provisions for energy conservation programs for low-income households.
II. Even the Playing Field by Providing More Resources to the PUC and Consumer Intervenors and Increasing Data Transparency (Governors, Legislatures, and PUCs)
Electricity rates are determined by proceedings called rate cases, in which utilities submit proposals and justifications to regulators, other intervenors (such as consumer advocates, environmental organizations, and state and local elected officials) submit testimony, and the regulators hold hearings and make a decision. Most rate cases end in settlement agreements between the utilities and other intervenors, facilitated by the PUC. Utilities drive this process—they file initial proposals and have more information about their system than other participants. Well-resourced PUCs and public interest intervenors are important to interrogate utility proposals and ensure that settlement agreements are a good deal for regular people.
In order to take on more responsibility in grid planning and utility oversight, PUCs need additional staff and analytical capacity. For example, a legislative commission in Texas found that the PUC needs more staff and resources to independently analyze utility sector data and provide sufficient oversight to ensure reliability. Funding for staff and analysis has a great return on investment—state leaders can save customers money and get better outcomes for a relatively small price.
Moreover, consumer advocate intervenors are typically underfunded compared to utilities and so cannot compete with utility proposals. This disparity in funding places consumer advocates in a position of exclusively reacting to utility requests, rather than having the bandwidth to interrogate existing system inequities or to develop potential innovative solutions to address ratepayer needs. Utilities also determine the pacing of their rate case applications, which can put consumer advocates even more on their heels. For example, in a 2019 rate case in Colorado, Xcel Energy brought 21 witnesses, while only a few consumer advocate intervenors testified.
Utilities in most states can recover the full costs of analysis and intervention legal fees from customers, giving the utilities significant resources to drive the process. States can prohibit this practice, directly reducing bills for customers and reducing utility influence over the process.
In addition to resources for PUCs and intervenors, data transparency can help even the playing field. Utility data are often difficult to access, embedded in filings that often run thousands of pages, and not standardized. This lack of data transparency makes it difficult for PUCs and consumer advocates to track utility spending and effectively intervene in rate cases.
Legislatures can provide additional funding for PUCs to hire additional staff and conduct independent analysis.
Legislatures and PUCs can prohibit utilities from recovering the costs of political activities from customers and limit the amount of legal fees that are recoverable from customers.
Legislatures can establish mechanisms to ensure that low‑income, consumer, and environmental justice advocates can participate meaningfully in PUC proceedings. Several U.S. states have implemented intervenor compensation programs or similar initiatives that reimburse reasonable costs for nonprofit organizations and community groups engaged in utility regulatory processes.
PUCs can charge utilities to fund independent analysis of utility proposals on behalf of customers.
PUCs can assess their processes with an eye toward reducing participation barriers for non-traditional docket participants, such as groups representing low-income or environmental justice communities.
PUCs can standardize reporting on utility costs and increase data transparency both during and in between rate cases.
Governors can direct agencies to conduct analysis to inform PUC proceedings and hire technical talent to engage with the PUC. Legislators can authorize and fund state agencies to conduct independent, proactive analysis to inform PUC proceedings, with opportunities for public input on the analysis.
California passed AB 1167 in 2025 that put an end to the use of ratepayer funds for political lobbying and strengthening enforcement against investor-owned utilities (IOUs) that illegally use ratepayer funds.
A 2023 Colorado law prohibited utilities from charging customers for lobbying expenses, political spending, trade association dues, and other similar activities.
In Illinois, a 2021 law expanded the Consumer Intervenor Compensation Fund to compensate consumer interest intervenors in planning and rate cases.
The Oregon PUC provides both Intervenor Funding and a dedicated Justice Funding program, supporting groups representing environmental justice communities and low‑income customers, with clearly defined funding caps for eligible participants.
Improve State Government Responsiveness to Clean Energy Projects
To build a clean energy project, developers must navigate a complex mix of state environmental permits, local and/or state siting approvals, and utility and grid operator interconnection processes. While federal permitting reform receives the most attention, most clean energy projects do not require federal approval and often get stuck due to state processes and local restrictions. For example, 73 percent of wind and solar projects that faced opposition in the 2010s were contested only at the state and local level, not federally.
In many places, these federal, state and local processes are not working for anybody—the clean energy developers who seek to build projects, the local communities in need of jobs and economic development, and the families and businesses struggling with rising utility bills.
What is going wrong? State agencies are often stretched thin, and outdated processes make it difficult to respond quickly to new projects. Most states lack a single designated authority at the state level who can oversee and enforce timelines. Permitting approvals often involve regional offices who take different approaches, increasing complexity and uncertainty for developers applying for permits. Inconsistent decision timelines increase risk for projects, raising costs.
In most states, local governments have control over siting, and each municipality has different processes and requirements, adding complexity for developers. Most states also lack a state authority that can ensure local governments do not unreasonably block or delay projects. About 20 percent of U.S. counties now have formal restrictions on clean energy projects.
State and local leaders can play a critical role in addressing these challenges. Governors can target agency capacity where it is most needed and legislatures can complement these efforts by funding permitting offices, creating statutory timelines to standardize reviews, and giving them enough decision-making power to actually meet those deadlines. State governments can leverage their capacity to evaluate costs, risks, and benefits—across timescales and geographies—of projects to inform decisions. Local governments can similarly improve and standardize their processes and support state implementation.
This brief focuses on a subset of solutions that help states be more responsive to clean energy projects. These solutions expand and improve government capacity to build clean power faster, lower utility bills, and demonstrate that the government can be an active and effective partner in solving problems.
Solutions
I. Increase Permitting Certainty and Consistency (Governors and Legislatures)
Uncertainty and inconsistency in permitting processes increases costs and delays projects. Timelines to hear back from an agency might vary considerably from one project to another, which adds uncertainty to project timelines. Some review processes are run by regional offices which may take different approaches to project evaluation (e.g., using different assumptions for modeling the impact of a project) and mitigation requirements. As a result, the same developer might go through a bespoke process for two very similar projects in different parts of the same state.
In addition, the scopes and goals of state environmental laws are often outdated. For example, environmental review often does not consider system-wide effects or second-order emissions impacts. As a result, environmental review often gets stuck on project-by-project analysis and lacks an overarching vision for the system.
States can address these issues minimal to no cost and without sacrificing the quality of environmental review by increasing certainty and consistency of permitting processes, centralizing capacity to run processes across agencies, and adjusting the goals and scope of environmental statute to include system-wide impacts and overarching climate goals.
Governors can issue clear guidance and standard operating procedures for analysis of impacts for different clean energy project types required under different laws (e.g., state clean water or environmental review statutes) and set timelines that agencies must follow for review and decisions.
Governors can set clear permitting goals for agencies and empower the “machinery” expertise (e.g., staff engineers, lawyers, environmental specialists, etc.) to meet those goals. Governors can also develop programs to upskill existing staff to expand the technical capacity needed for permitting operations.
Governors can issue guidance on mitigation requirements that projects can take to shorten review.
Legislatures can also set statutory decision timelines and limitations on what impacts are considered to further increase certainty for projects. In some cases, legislatures may need to provide agencies with the authority and resources required to standardize mitigation requirements and speed up timelines.
Legislatures can re-align the goals and requirements of permitting statutes and environmental review to prioritize system-wide goals.
Legislatures and governors can exempt certain clean energy projects from state environmental permitting or create simplified permitting pathways for such projects.
Legislatures can mandate and support adoption of instant digital permitting for distributed energy resources located at homes and businesses, drastically reducing the cost to install rooftop solar and energy storage.
Governors and legislatures can consolidate agency authorities, reducing the number of process steps for developers and conflicts between agencies.
In Pennsylvania, solar developers must obtain a stormwater permit from the Department of Environmental Protection (DEP), a process that involves working with regional conservation districts on stormwater analysis and agreement on mitigation requirements. Developers have struggled with inconsistent approaches among conservation districts on modeling assumptions and mitigation requirements. In December 2025, DEP issued updated its Solar Panel Farms Frequently Asked Questions to clarify key analytical inputs for solar developers and will increase consistency in conservation district approaches.
The New York State Legislature passed the Accelerated Renewable Energy Growth and Community Benefit Act, mandating specific timelines for permit reviews, consolidating authority across agencies, and providing funding to support dedicated staff. This statutory framework has significantly reduced approval times for large-scale renewable projects.
Pennsylvania Governor Shapiro also directed state agencies to evaluate the timelines for each permitting process and set a specified maximum timeline for eligible projects by which applications will be processed.
In 2025, state legislatures in New Jersey, Texas, and Florida passed laws requiring local governments to adopt instant digital permitting processes for distributed resources.
Arizona House Bill 2003 allows utilities to replace conductors or structures on an existing transmission line without needing to apply for a Certificate of Environmental Compatibility (CEC) as long the line has a valid CEC (or has been grandfathered in) and all original conditions continue to be met.
In Minnesota, HF 4700 consolidated certain permitting responsibilities into a single law and shortened the timeline for state regulators to review and permit clean energy projects.
Texas passed SB 20 in 2005, establishing Competitive Renewable Energy Zones that smooth the process of developing and integrating renewable energy projects into the grid. This program has helped bring more than 18 GW of wind capacity online.
II. Increase Siting Certainty and Consistency (Governors, Legislatures, Local Leaders)
In most states, local governments handle siting of clean energy projects. Municipalities and counties within the same state may take wildly different approaches to siting, including different fee structures, setback requirements, public input requirements, approval processes and timelines, etc. This variability makes it harder to build clean energy projects without added benefit to communities. State leaders can improve certainty and consistency without sacrificing project quality or local benefits.
Legislatures can standardize processes across local governments, including by setting standard timelines for decisions, prohibiting excessive restrictions on clean energy projects and grid upgrades, and limiting the reasons for which a project can be denied approval.
Legislatures can move siting authority to the state level for large projects or projects that meet certain criteria (e.g., projects that create good jobs and economic benefits).
State lawmakers can create a streamlined, one-stop permitting process for distributed energy resources. This process would consolidate building, zoning, and environmental approvals. Such a framework reduces delays, lowers costs, and provides developers and homeowners with greater certainty.
Governors can develop model siting ordinances and encourage or incentivize local governments to adopt them.
Local governments can voluntarily work together to align processes.
Colorado developed a clean electricity code repository that included principles for smart local code design for clean electricity deployment.
Michigan HB 5120 financially incentivizes local governments to avoid overly restrictive ordinances and creates a state-led pathway for projects to by-pass overly restrictive local ordinances.
Arizona House Bill 2019 standardized timelines for municipalities to make permit decisions and took steps to ensure municipalities are responsive, including requirements on providing contact information and communicating with permit applicants.
A bill introduced in the Oregon legislature would exempt clean energy projects eligible for soon-to-expire federal tax credits from the state’s onerous state siting process.
Hawaii’s proposed SB588 creates a self-certification and standardized permitting system for behind-the-meter solar and storage projects.
Illinois HB 4412 set statewide renewable energy project siting standards that takes supremacy over unduly restrictive local decisions.
Indiana’s SB 411 set voluntary siting standards for wind and solar power projects. If communities meet those standards, they’re pre-cleared for project development.
New Mexico’s Renewable Energy Transmission Authority established an MOU with the Federal Permitting Improvement Steering Council to improve coordination and receive assistance on eligible permitting projects.
III. Create Dynamic Agencies that Can Respond to Project Needs (Governors and Legislatures)
States should build dynamic, flexible agencies that are responsive to evolving barriers to clean energy deployment. Government responsiveness is often thwarted by limited or outdated information, poor information sharing across agencies, and lack of centralized decision-making. Governors and state agency leads often do not know which specific processes are causing delays or uncertainty for projects. Where agencies have that information, it is often not shared among agencies or with the governor’s office without intervention. Agencies often do not have the mandate or the capacity to track and share this information.
A mandate and resources to collect detailed information on project barriers, and to share that information across agencies, allows states to be more responsive to clean energy industry needs. Using this information, states can ensure that limited agency capacity is targeted where it can have the greatest impact and helps agencies anticipate, rather than react to, common obstacles.
Governors can identify the specific government processes and other barriers that are holding up projects and increasing uncertainty through direct engagement with developers and formal processes like Requests for Informations. Governors can survey developers for specific information on the failure points in the process of getting projects built, across permitting, siting, interconnection, etc. Governors can then address the critical bottlenecks by adjusting the processes that are outdated or not serving the public or shifting capacity where processes must be maintained but are moving too slowly.
Governors can build centralized processes—for example by designating a senior official as state-wide lead for permitting coordination—that can quickly respond to bottlenecks and share information across agencies. Governors and legislatures can designate a single agency as the lead on permitting, siting, and project assistance and provide that agency with the authority to make decisions. For these steps to work, governors must provide the lead official or agency with the adequate decision-making authority and capacity to run the process.
Legislators can provide the resources for the above-referenced information collection and the authority for centralized permitting processes.
The Colorado Energy Office and Department of Natural Resources conducted an extensive survey of developers, local governments, community organizations, and other stakeholders to detail clean energy siting and permitting issues and develop a plan to address them.
Following passage of HR 1 in Congress, which rapidly phased out tax incentives for certain renewable energy technologies, Governors’ offices in states like North Carolina and Pennsylvania organized inter-agency dialogue with private sector stakeholders to identify and accelerate projects that could form cost-saving federal incentives before they expire.
The California Energy Commission runs a consolidated permitting process that centralizes staff capacity to manage permitting, with specified review timelines for clean energy projects that meet certain job quality and community benefits criteria.
Washington House Bill 1216 authorized the Department of Ecology to run a consolidated permitting process to collect all relevant information from developers and coordinate across agencies to speed up review. That bill also created an interagency Clean Energy Siting Council to improve how projects are sited in the state.
In New York, Governor Kathy Hochul directed state agencies to accelerate renewable project approvals by reviewing and reforming agency processes to reduce backlogs and coordinate efforts across departments, from environmental review to project eligibility screening, focusing agency attention on projects ready to leverage expiring federal tax incentives.
IV. Speed Up Interconnection Timelines (Governors, Legislatures, and Public Utility Commissions)
Long timelines for approvals to connect to the grid and to complete necessary transmission upgrades are one of the largest drivers of project cancellations and delays. States have little control over the interconnection process for projects connecting to the bulk transmission system, which is regulated by the Federal Energy Regulatory Commission. However, states can help projects use surplus interconnection processes, which enable faster approval for projects that are using excess interconnection capacity at existing generators. States also have control over the interconnection process to connect smaller projects to the distribution grid, which is run by utilities and regulated by the PUC.
Governors can direct state energy offices proactively identify and map surplus interconnection capacity and use state procurement authority, expedited permitting processes, and incentives to encourage clean energy development at those sites. This analysis should also include sites that may have excess interconnection capacity because projects (generators or new large load sources) fell through or shut down after triggering grid upgrades
Legislatures can require PUCs and utilities to consider using sites with underutilized transmission (e.g. sites of peaking gas-fired power plants that operate very infrequently) for clean energy development.
Governors and PUCs can identify zones where load is likely to increase (e.g., due to data center deployment, electrification of ports, and heavy-duty freight trucking) and fast-track distributed interconnection for projects in those areas.
PUCs can require that utilities develop rapid procurements for clean energy resources at sites with surplus interconnection.
In California, AB 1408 would have required state agencies, the state grid operator, and utilities to incorporate surplus interconnection capacity into long-term planning, but it was vetoed by Governor Newsom.
Virginia HB 1065, introduced in the 2026 legislative session, would task the State Corporation Commission with conducting a study of available surplus interconnection potential, and requires the state’s largest utility to procure new capacity via surplus interconnection.
In Indiana, HB 240, proposed in 2026, would require utilities in their integrated resources plan to analyze how much extra interconnection capacity they have and how they can use that existing capacity to bring more generation resources online. And, it would require such analysis to be done before permitting new power plants.
V. Provide Additional Funding for Siting and Permitting (Governors and Legislatures)
Clean energy permitting delays often stem from understaffed or under-resourced agencies struggling with outdated processes and technology. Legislatures can address this by increasing dedicated funding for permitting offices, enabling agencies to hire technical staff, invest in modern permitting platforms, and reduce backlogs. Allocating funding for targeted reforms can substantially shorten approval timelines. Potential examples of such include digital permit tracking systems, programmatic environmental reviews for common project types, and training specialized clean energy reviewers. States can start by digitizing permitting processes and making it easier for projects to submit the required information. States can then collect information from developers on the processes that are causing the most difficulty and provide targeted staffing and resources to address these bottlenecks.
Governors and legislatures can digitalize state permitting processes to improve transparency and interagency workflow management.
Governors and legislatures can institute state-level digital permitting platforms.
Governors and legislatures can create shared staff resources to hire the high-demand technical expertise necessary for permitting and enable those experts to move quickly between agencies in response to need.
State agencies and legislatures can initiate programmatic environmental reviews for clean energy projects subject to state environmental review laws.
Virginia’s Permitting Enhancement and Evaluation Platform (PEEP), now expanded as the Virginia Permit Transparency (VPT) system, was enacted by executive order. The VPT provides a public dashboard to track permits through state processes and workflow management tools for agency staff.
The Washington State Legislature created the Clean Energy Siting and Permitting (CESP) Grants Program, providing roughly $4.85 million to local governments, tribes, and state agencies to hire staff and modernize permitting workflows for solar, wind, and storage projects. By funding technical staff and process improvements, the legislature helped agencies reduce backlogs and provide more predictable permitting timelines for developers.
In 2025, Washington’s Department of Ecology, under direction from the legislature, completed programmatic environmental reviews for clean energy projects to speed permitting application and review of onshore wind, solar, and hydrogen projects in the state.
In Arizona, digitization of several environmental permitting processes helped reduce decision timeframes by 91 percent.
Build Administrative Capacity to Plan for an Affordable & Reliable Grid
Today, the U.S. bulk transmission system faces significant constraints that limit where new clean energy projects can be built and threaten overall grid reliability. Many regions with abundant clean energy resources simply do not have enough high-voltage transmission capacity to deliver that power to population centers. As a result, developers are increasingly unable to move generation projects forward even when siting, permitting, financing, and interconnection queue positions are in place. Without new transmission capacity, interconnection backlogs grow, power costs increase, and states are forced to rely on older fossil resources simply because they are already in place.
Transmission buildout is thwarted by barriers such as long planning timelines of 7 to 15 years, route identification, environmental review, litigation, supply chain constraints, and fragmented and inadequate planning processes.
While the permitting reforms described elsewhere in the playbook would help, we won’t build the transmission system that we need without improved planning. Building transmission lines requires utilities, developers, customers, and grid operators to work together to determine where a transmission line is needed and appropriately allocate costs across different stakeholders. Without a strong administrative state that can facilitate the process and collect and share all the required information (such as congestion on current lines, hotspots of demand growth, areas with high potential for cheap clean energy, etc.), this process often fails and very rarely results in optimal expansion of the transmission system. Today, states and grid operators lack administrative capacity to conduct this planning process, which is hamstringing our ability to expand the grid.
States can build the capacity to improve planning in order to spur development of transmission lines with the greatest benefit for the public.
Solutions
I. Include Advanced Transmission Technologies in Planning (Legislatures, Governors, and Public Utility Commissions)
Advanced Transmission Technologies (ATTs) can be used to increase grid capacity on current rights-of-way, alleviating congestion and allowing for more efficient energy transfer without building new infrastructure. Utilities being able to increase efficiency and cost effectiveness of their infrastructure is especially important as load growth continues to increase across the country and raise retail electricity bills. For example, installing high-performance conductors increases the amount of electricity that can be transferred over an existing transmission line. By one estimate, reconductoring with these technologies could double transmission capacity on the current grid. Dynamic line ratings allow lines to carry more electricity when weather conditions are good, rather than defaulting to conservative limits on line capacity. Each type of ATT has its own advantages and benefits.
PUCs can dictate standards, enforce rules, conduct studies, and establish new policies that require and incentivize utilities to evaluate and deploy ATTs.
Legislatures can require utilities to include evaluation of ATTs in planning processes, conduct studies on ATT potential and deployment opportunities, and analyze ATTs as potential enhancements to new transmission infrastructure.
Governors can petition ATT rulemakings to the PUC via an executive order, can integrate ATTs into funding criteria for grid or resilience projects and direct economic development agencies to study the economic impacts of ATTs, and convene ATT task forces to set direction and collaborate with educational institutions to develop workforce training programs focused on ATTs installation, operation, and maintenance.
Utah’s SB 191 requires utilities to conduct an alternatives analysis for ATTs in IRPs and also provides language that the Commission can approve cost-recovery for ATTs if it is determined the deployment is cost-effective.
Ohio’s HB 15 requires that utilities summarize ATT evaluation in power siting board certificates and furnish annual 5-year reports on ATT deployment opportunities, including congestion mitigation studies and that the PUC evaluate the potential of ATT deployment including consultation from stakeholders via two public workshops.
Governor Wes Moore’s December 2025 Executive Order Building an Affordable and Reliable Energy Future creates a Transmission Modernization Working Group that makes ATT policy recommendations to the Maryland Energy Administration, which in turn makes formal petitions to the PUC.
Montana House Bill 729, adopted in 2023, enables the state PUC to set cost-effectiveness criteria to allow utilities to deploy advanced transmission conductor technologies and recoup the cost via their ratepayers, similar to investments in new energy generation.
II. Create a New Transmission Planning Authority (Governors and Legislatures)
Lack of coordination between transmission and generation planning creates inefficiencies and prevents smart clean energy development. In deregulated markets—and in some vertically integrated states—transmission and generation planning processes occur largely in isolation without systematic processes to align long-term clean energy expansion with major grid upgrades. While the federal government has authority to set the rules for planning regional and interregional transmission lines, state leaders have tools at their disposal to expand transmission buildout and improve planning.
Legislatures can create transmission planning authorities explicitly authorized to identify transmission corridors that can expand low-cost clean energy generation, lead on the permitting and siting of transmission lines, secure project finance, negotiate and collaborate with other states on interstate transmission plans, provide advice on transmission priorities and planning needs for the state, and enter into public or private partnerships to help with project development. These authorities must be empowered and resourced to collect all the necessary information (e.g., congestion on the existing system, load forecasts, sites of cheap clean energy, etc.) and to attract top talent with expertise in utility planning, project development, and financing.
Governors can create a transmission advisory or coordinating committee and reorganize state agencies, boards, and commissions to serve the purpose of a transmission authority or to create one.
New Mexico passed the Renewable Energy Transmission Authority (RETA) Act in 2007, creating RETA and authorizing it to “plan, license, finance, develop and acquire high-voltage transmission lines and storage projects to help diversify New Mexico’s economy through the development of renewable energy resources.”
In Colorado, SB21-072 created the Colorado Electric Transmission Authority (CETA) to plan and develop transmission lines to increase reliability and deploy more clean energy. CETA has very similar powers to New Mexico’s authority.
III. Require Integrated Transmission and Generation Planning (Governors, Legislatures, and Public Utility Commissions)
Coordinated planning is essential to ensure that transmission is expanded in the right places and that new clean energy investments can flow to areas with sufficient transmission capacity. Around 35 states require their utilities to develop Integrated Resource Plans (IRPs), which act as a roadmap for how the utility will meet future forecasted electricity demand over a specific time period. Although transmission and generation are key inputs for energy supply, they are usually not included in these plans. The result is piecemeal grid planning, as transmission providers and developers focus on smaller lines which meet near-term needs and are profitable within their own footprint.This shortcoming is a product of both process—regulators and state agencies have not been mandated to link transmission and generation planning—and capacity, where the administrative state lacks the right staff and resources to conduct integrated planning.
Integrating these processes can ensure better coordination between load and generator interconnection, a more holistic understanding and roadmap of current and future grid reliability and supply chain needs, help avoid duplicative investments and ensure costs for upgrades remain reasonable, and can lower the likelihood of stranded or undersized assets. This integrated planning is especially important in places with projected load growth, whether from data center buildout or electrification of buildings, heavy-duty transportation, or factories.
Governors can direct relevant agencies to work with grid operators, PUCs, and utilities to encourage integrated planning.
Legislatures in vertically integrated states can require utilities to conduct IRPs where they don’t already do so and further require generation and transmission planning to be integrated.
PUCs can require utilities to link transmission and generation planning.
Enacted in 2021, Nevada S.B.448 requires an electric utility to amend its most recently filed resource plan to include a plan for certain high-voltage transmission infrastructure construction projects that will be placed into service before 2029.
In 2022, a Memorandum of Understanding (MOU) between the California Independent System Operator (California ISO), the California Public Utilities Commission (CPUC), and the California Energy Commission (CEC) ensured that the planning and implementation of new transmission and other resources were linked, synchronized, and transparent.
IV. Ensure Effective Implementation of FERC Order 1920 (Governors, Legislators, and Public Utility Commissions)
Recent federal actions, such as FERC Order 1920, have the potential to be a useful tool for states if implemented correctly and efficiently. FERC Order 1920 requires long-term, forward-looking, multi-value regional planning. It was designed to improve transparency in local transmission planning, including by conducting local stakeholder meetings. Under this filing, transmission providers must produce long-term, at least 20-year, regional transmission plans at least every five years, which must utilize seven specific categories of forward looking factors, select projects based on different economic and reliability benefits, and consider the use of grid-enhancing technologies.
Governors can take a more active role with PUCs to guide their involvement in regional transmission planning processes established under FERC Order No. 1920.
State legislators can hold hearings with PUCs on how utilities, regional transmission planners, and state officials plan to participate and support regional planning and put the order into action.
In the mid-Atlantic, 69 legislators from 10 states called on PJM to implement FERC Order 1920 without delay due to the benefits of reliable, affordable and clean electricity it will bring to their constituents.
Fix Broken Incentives to Expand Distributed Energy Resources
Misaligned incentives for utilities have limited the types of technologies and solutions in play to meet demand growth and maintain reliability.
Distributed energy and grid flexibility solutions—such as rooftop solar, flexible demand, smart charging for electric vehicles, and distributed storage, as well as alternative transmission technologies—can help meet demand growth faster and cheaper than solely relying on large power plants and bulk transmission upgrades. Distributed energy resources are particularly useful in an era when interconnection delays and economic pressures on rate payers have slowed down efforts to build large-scale transmission projects and generation facilities.
Investor-owned utilities make a profit based on a fixed rate of return on certain expenditures. Utilities typically do not earn a return on distributed energy resources and grid flexibility programs so many utilities have underinvested in these solutions and underutilized them for grid planning.
Largely because of these broken incentives, there’s a lack of capacity to value, procure, and orchestrate these distributed resources, thus limiting their ability to scale as a utility resource. In most states, utilities and regulators do not consider distributed resources in planning processes. Where distributed resources are considered, it is often only through voluntary offerings and limited pilot programs.
Leveraging distributed resource solutions requires state leaders to fix broken incentives and build new government capacity tools to facilitate uptake and utilization of these technologies.
Solutions
I. Incentivize adoption and use of distributed energy resources (Legislatures and Public Utility Commissions)
States can correct misaligned incentives by creating mechanisms to value the benefits of distributed energy resources and incentivize optimal utilization of distributed resources to reduce spending on new generation and grid upgrades.
Legislatures can establish Virtual Power Plant programs—run by utilities, public developers, or partnerships between public developers and utilities—to aggregate distributed energy resources to best serve the grid and compensate these resources accordingly.
Legislatures can incentivize adoption of appliances and customer energy systems, like smart chargers and thermostats, capable of supporting the electricity grid.
Legislatures and PUCs can require utilities to create mechanisms to value the benefits of distributed resources and adjust profit motives through performance-based ratemaking to align utility incentives with expanding distributed resources.
In Colorado, SB24-218 required Xcel Energy to create a new mechanism to compensate customers for distributed energy resources that can benefit the grid.
In New Jersey, Governor Sherill issued an Executive Order directing the PUC to establish a Virtual Power Plant program to aggregate distributed energy resources and use them to benefit the grid.
II. Require procurement of distributed resources (Governors, Legislatures, and Public Utility Commissions)
Utilities generally do not procure distributed resources, focusing instead on procurement and buildout of large-scale generation. Formal procurement of distributed resources can maximize use of the existing grid and get new generation onto the grid fast, which can avoid spending on the distribution grid and save customers money.
Legislatures can set statutory requirements for minimum procurement amounts.
PUCs can require utilities to conduct procurement of distributed clean energy.
Governors can direct PUCs to initiate solicitations for distributed clean energy and set timeframes and minimum procurement amounts that PUCs and utilities must meet.
In Minnesota, Xcel Energy has proposed a Distributed Capacity Procurement of 50-200 MW of distributed energy storage. Under the procurement, the utility would contract with a developer to build small-scale batteries and pay households, businesses, and non-profit organizations to host them.
In Illinois, the Clean and Reliable Grid Affordability Act required the procurement of 3 GW of storage and created a mechanism to compensate households and businesses for customer-owned storage. The state has a government agency—the Illinois Power Agency—that develops procurement plans and runs competitive procurement processes.
In Maine, the PUC issued a Request for Proposals for solar projects on PFAS-contaminated farm land.
In New Jersey, Governor Sherill directed the PUC to procure an additional 3,000 MW of community solar through the NJ Community Solar Energy Program.
III. Improve interconnection processes for distributed resources (Governors and Public Utility Commissions)
The interconnection process is a major hurdle for new clean energy projects. Utilities run the process to connect projects to the distribution grid, and PUCs regulate this process. Because utilities typically do not profit from distributed generation, they have generally deprioritized improvements to the interconnection process for distributed generation. PUCs have not had the mandate to push utilities on improving the interconnection process for distributed resources, which means capacity has not been channeled toward this goal. State leaders can force utilities to speed up the interconnection queue to get more clean energy on the grid.
Governors can direct PUCs to improve the interconnection process with specific goals (e.g., reduce the application approval time by a specified amount) and penalties for non-compliance.
PUCs can work with utilities to make improvements to the interconnection process to shorten timelines and provide more certainty to developers. PUCs can dedicate staff resources to this issue to ensure rapid improvements to the process.
Colorado Governor Polis in an August 2025 directive tasked state agencies to pursue flexible interconnection and voluntary curtailment for distributed energy and community solar projects; work to facilitate the pre-purchase of project equipment and/or affiliated electric transmission and distribution infrastructure; and to pursue updates to interconnection standards for customer-sited projects, including the use of meter collar adapters and other measures to minimize the cost and time. The Colorado PUC is now working to allow projects to use flexible interconnection agreements—alowing projects to connect to the grid in constrained places if they agree to use technologies that can operate flexibly and avoid straining the grid—which can speed up timelines and reduce interconnection costs.
In New Jersey, Governor Sherrill directed the PUC to take several steps to standardize and speed up interconnection of clean energy projects to the distribution grid.
IV. Improve data and analysis and increase transparency to optimize deployment (Governors, Legislatures, and Public Utility Commissions)
Expanding distributed energy resources in the right places is key to maximizing benefits to the grid and reducing costs for customers. States can help inform strategic adoption through data collection and analysis on the optimal places to deploy customer-owned resources and increase transparency of those data for the public.
Policymakers can analyze opportunities that will make the most impact through avoided costs of updating the grid or building bulk generation. Distributed energy projects are most useful when they minimize constraints on the distribution system and reduce peak demand. Information sharing can be facilitated through either Integration Capacity Analysis or regular Integrated Distribution Planning.
State leaders can also identify potential projects to provide tailored support by engaging developers and site hosts directly through Requests for Information. States can then use this information to direct support to projects. For example, data collection can enable aggregation and standardization of information about siting readiness, interconnection considerations, terms and conditions, and resilience needs which can then be converted into a pre-qualified inventory for procurement or financing.
Governors can direct agencies to engage with developers and other stakeholders to identify barriers to getting distributed projects built and identify solutions to support projects (e.g., connecting host sites with developers and financiers).
Governors and PUCs can improve distribution system planning to better anticipate infrastructure needs and ensure that distribution system planning is synced with grid operator interconnection processes and transmission planning.
PUCs can require utilities to conduct and publish analysis on hosting capacity and locational value of distributed clean energy resources and establish a market for distributed energy resources.
In Massachusetts in 2024, the PUC directed utilities to incorporate “non-wires alternatives”, including distributed energy resources, into system planning. Now, one of the state’s largest utilities has launched an innovative mechanism to quickly procure distributed energy resources that provide the greatest costs reductions and grid benefits.
The New York Build‑Ready program identifies potential host sites and shifts early development duties (i.e., negotiate the initial lease and start the interconnection process) from private developers to the state, which accelerates the move of renewable energy projects from the idea stage to construction.
In New Jersey, Governor Sherill directed the PUC to work with the utility to improve hosting capacity maps for distributed energy resources.
V. Create a Municipality or Sustainable Energy Utility in lieu of an Investor Owned Utility (Legislatures and Local Leaders)
Creating a municipal utility or sustainable energy utility (SEUs) can address bypass misaligned incentives and focus on the technologies and strategies that most benefit the public. These entities differ from traditional investor owned utilities because they are not-for-profit, owned by the communities they serve, and are run by local government. Benefits of this utility model include securing more affordable electric rates for consumers, achieving renewable and clean energy goals more quickly, increasing local control and governance over energy decisions and infrastructure, and contributing to local economic development.
Legislators and local leaders can spearhead the effort of municipalizing by developing a concept and authorizing or establishing a municipal utility. State and local leaders can explore different options, including developing a supplemental utility that procures generation but still uses the private utility’s poles and wires or fully acquiring the private utility and creating a public entity to run the full system.
In 2024, residents of Ann Arbor, Michigan voted to authorize Proposal A: Creation of a Sustainable Energy Utility. The Sustainable Energy Utility (SEU) will be an opt-in, supplemental, community-owned energy utility that provides 100% renewable energy from local solar and battery storage systems.
In 2005, residents of Winter Park, Florida voted to authorize the city’s use of bonds to buy the local distribution facilities from the incumbent utility.
The state of Nebraska receives all of its electricity from publicly owned sources.
Wield Creative Finance Tools to Drive Investment and Reduce Capital Costs
Rollbacks of federal financial support have threatened the viability of many clean energy projects. State and local leaders can help keep projects alive and build new ones with creative financing tools. In some cases, this means taking a more active role in coordinating across public and private sector actors, while in others that means building entirely new administrative capacities to perform more ambitious financial transactions or act as a public developer.
In addition, the grid is facing new challenges that require massive investments. For example, recovery from and preparation for wildfires is inflating energy bills in the west. Gulf states are facing similar costs from hurricanes. States need creative finance tools to ensure that these costs do not continue to raise bills for regular people and small businesses.
Beyond merely acting as a source of capital, governments of every shape and size actively participate at every stage in the project development and planning lifecycle to bring down the total cost of projects. These include lowering financing costs, securing stable or catalytic financing, and providing an avenue to complement other functions the state is undertaking. Local governments can engage in public development functions, including through creative finance tools and engagement with community choice aggregators, rural electric cooperatives, and energy service companies.
Solutions
I. Empower development entities with the legal authority and staffing to pursue high priority projects (Governors, Legislatures, Local Leaders)
State leaders can help ensure that infrastructure authorities, city and county development corporations, or energy departments of a given jurisdiction have the relevant borrowing authority, ownership and operation powers, and partnerships capabilities to support project development.
To be successful, state financing entities or public developers need clarity and certainty on how projects they support can participate in electricity market operations, including whether projects can participate in utility procurement processes or interact with grid operator interconnection processes. State financing also must be coordinated with other grid planning processes.
Given the overlapping interests state and local economic development agencies may hold, this process will demand adequate staffing resources and may require significant stakeholder engagement with private sector actors, government officials, and others.
Legislators can write or amend enabling authorities to explicitly provide state and local entities with the financing, bonding, ownership, and partnership authorities necessary to support, finance, own, and/or operate projects. These authorities should include co-financing and co-development options to blend public and private support. Legislators can also make sure that these authorities are flexible and broad so that state development can be competitive with private developers.
Legislators can allow use of public financing tools to support certain projects. In particular, legislators can expand the bonding authority available to state agencies for use on clean energy projects.
Legislators can establish state and/or utility procurement targets for clean energy, storage, and grid projects and provide direction and clarity for state financing entities to service these procurements.
Governors can use their authority over appointments and interagency coordination to align disparate entities around specific tangible objectives.
Governors can draw on recent public private partnerships in the offshore wind industry to structure offtake, procurement, and other commercial activities with utilities and developers across a range of clean energy projects. State entities can seed virtual power plants, solar, wind, energy storage and other clean power projects that mutually derisk projects for both public and private developers alike.
Governors and legislators can provide expedited permitting and siting processes for publicly sponsored projects.
City and county officials can form project-specific entities or special purpose authorities to make projects financeable.
In New Mexico, the Renewable Energy Transmission Authority (RETA) was established in 2007 and was granted statutory power to exercise eminent domain to acquire property or rights of way for eligible renewable energy projects. This authority has been critical in overcoming fragmented land acquisition barriers.
The Connecticut Green Bank’s Solar Marketplace Assistance Program (Solar MAP) serves as a public developer to finance and build solar projects for K-12 schools, allowing the state to own the assets and sell power back to districts at a discount. While the Green Bank has acted as a public public developer in some form since 2014, projects from Solar MAP are projected to deliver tens of millions of dollars in savings all without incurring any upfront costs for districts.
In Colorado, SB 21-072 in 2021 created the Colorado Electric Transmission Authority as a special-purpose development authority granted power to issue bonds and corridor acquisition tools.
II. Use pooled loan funds like state bond banks to lower borrowing costs and build project pipelines (Governors, Legislatures, and Local Leaders)
Pooled borrowing authorities offer transaction efficiency and credit strength for cities, counties or small utilities paying the fixed costs of a standalone bond issuance by aggregating relatively modest projects into standardized pools. This reduces the issuance and underwriting costs, and can often enhance credit resulting in lower borrowing costs.
Bond banks are valuable in practice because they are a repeatable financing infrastructure that can be improved and expanded over time. Governors offices, county executives, and mayors can direct agencies to build a steady pipeline of eligible projects (using Requests for Information or direct engagement) and then work with relevant financing authorities to standardize project intake, selection, and reporting and make the whole process more repeatable.
Once local governments experience lower borrowing costs and faster execution through a standardized conduit, the model becomes politically sticky and easier to scale, especially when paired with complementary tools like revolving funds or credit enhancement that can serve smaller borrowers and accelerate project turnover.
Legislators in states that lack a bond bank can establish one capable of pooling local loans, issuing bonds, and relending the proceeds. They can further work to standardize project solicitation, underwriting, and closing cycles to ensure the institution creates a regular cadence.
Legislators in states that have a bond bank can expand eligible project types (to include clean energy projects and resilience priorities like building retrofits, microgrids, etc.) and create standard project templates.
Governors can work with state agencies to centralize the origination of bonds for a public developer in their state’s bond bank or otherwise help public developers and other financing agencies exercise their bonding authority.
Governors can make regular use of bond banking authority a priority by directing agencies to run a standing intake process, and appoint or empower relevant state personnel to highlight pooled lending as an innovative solution.
City and county officials can create a rolling inventory of eligible projects, bundle them into multi-jurisdiction project aggregators and engage with existing bond banks on technical assistance for project scoping and diligence.
Vermont’s Bond Bank issues bonds backed by repayments of its loans to individual municipalities, school districts, etc. and maintains a dedicated Municipal Climate Recovery Fund. The bank has the ability to backstop non-payment by municipal or county entities that fail to pay based on state funds allocated to municipal or district borrowers in what is known as an “intercept mechanism.”
Virginia Resource Authority’s Resilient Virginia Revolving Fund was established in 2022. Jointly administered with the state’s Department of Conservation and Recreation, the pooled borrowing platform provides financial assistance for flood-mitigation projects across the state.
III. Require energy utilities to supplement portions of their debt or equity with public bonds (Governors, Legislatures, and Local Leaders)
A unique characteristic of public development is that strategic capital deployment has the potential to derisk private investment. Mandating that utilities replace a portion of their high-cost equity with state backed public debt or revenue bonds optimizes the project’s capital stack, thereby reducing the average cost of capital and reducing the total financing costs for capital-intensive grid infrastructure. Investor-owned utilities typically finance large infrastructure projects through a mix of debt and equity with regulators guaranteeing a return on equity (ROE) to attract private investors. Because this ROE is significantly higher than the interest rates on public debt, requiring public bonds to supplement the capital stack can dramatically reduce the long-term costs that are ultimately passed on to ratepayers.
This mechanism leverages the state’s superior credit rating and tax-exempt status to fund the most expensive portions of development while leaving the utility to focus on its core competencies of construction and grid operation. Establishing a public financing facility in this way allows the public sector to act as a sponsor investor for projects of high public interest, such as interregional transmission lines. By providing lower cost debt, states can ensure that critical energy targets are met without placing an undue financial burden on households. This approach creates a more stable investment environment and allocates risks more effectively across public and private stakeholders.
Legislators can mandate investor-owned utilities make use of state-backed revenue bonds or other forms of public debt to finance high-priority capital investments such as grid resilience or interregional transmission.
Legislators can authorize state infrastructure banks or other financing authorities to act as sponsor investors and displace high cost equity of a project’s capital.
Governors can establish dedicated clean energy project finance working groups to examine the full scope of infrastructure financing tools needed to derisk capital investment in transmission, generation, distribution and other electricity assets.
Regulators and state energy offices can lower the costs passed along to ratepayers by integrating public financing facilities directly into RFP processes, allowing bidders to access lower-cost capital.
City and county officials can pass local resolutions advocating for a specific local utility project to be financed via public bond rather than traditional utility equity to ensure the lowest possible rate impact for their residents. A similar strategy can be pursued via written submission or intervention within PUC docket proceedings.
City and county officials can collaborate with state energy offices to identify projects that are ideal candidates for public debt supplements.
California’s 2025 law SB 254 establishes a state public financing facility (the Transmission Infrastructure Accelerator) to replace high-cost utility equity with lower-cost public debt for new transmission projects, directly reducing the ratepayer impact of CAISO’s multi-decade development plans. The law requires utilities to finance billions of dollars of grid hardening investments using bonds instead of utility equity financing, reducing costs for customers and preventing the utility from excessively profiting off of this set of expenditures.
Maine’s Clean Energy Financing Study recommends operationalizing state revenue bond authority and establishing a working group on large clean energy project finance to optimize the capital stack for clean energy and transmission projects.
IV. Develop greater public understanding about the development levers available to public or quasi-public entities (Governors, Legislatures, and Local Leaders)
Some financial functions like loan issuance, co-financing, and non-dilutive debt financing may be well known to state energy offices, green banks, and certain infrastructure authorities. But in general, public financing is hampered by a lack of clarity, information, and standardization of different agencies’ authorities.
States can maximize the impact of public resources by establishing clear financing authorities and responsibilities, providing state authorities with broad powers to flexibly support projects, ensure that public finance is prioritizing the right investments, and providing clear direction on how publicly sponsored projects support utility procurement or grid operator processes. In addition, standardizing state and local financing entities drives down costs by making processes more repeatable and can pave the way for more effective federal support in the future. By surfacing all the capabilities public entities currently have and may wish to develop in the future, policymakers and advocates can align on objectives to strengthen the public developer toolkit and bring clean energy projects closer to fruition.
Governors can inventory borrowing, contracting, and financing authorities and provide clear guidance on roles and responsibilities between agencies.
Governors and legislatures can require reporting on key performance metrics like deal volume, borrower participation, and time-to-close to help encourage institutionalization.
Governors and legislators can publish analysis and information on areas to focus energy project development and create special zones for the installation, procurement, manufacturing, or operation of energy projects of various kinds. These industrial zones could provide access to a variety of benefits: expedited permitting, siting, interconnection, specific public finance facilities, funds for resiliency + operation, and various other coordination benefits from other interested state agencies.
Legislatures can provide agencies with clear financing authorities, direction on what types of projects to support, and a broad set of tools to flexibly support projects.
City and county officials can examine if there are relevant state laws that require additional ordinance/resolution to use. Some tools to activate and then specify rules to create repeatable administrative playbooks.
Houston, Texas had to pass an authorizing city ordinance to activate a state program known as Property Assessed Clean Energy (PACE). The program allows commercial and multifamily property owners to finance energy efficiency, renewable energy, and water conservation improvements and has invested over $540 million dollars statewide since its inception in 2016.
Montgomery County, Maryland created a green bank in 2016. In 2022, the county passed a statute to direct 10% of the county’s fuel tax revenue to the Montgomery County Green Bank each year. The green bank completed a new bus depot for EV buses in 2022 co-located with a 6.5 MW microgrid that can run independent of the local utility.
Colorado has an EPC program that lends against a project’s anticipated cost savings to finance building retrofits.
Protecting America’s S&T Ecosystem
Over the past eighty years, the United States has maintained both economic and military primacy thanks to our technological superiority. Other countries have sought to replicate this advantage, some of whom (particularly the People’s Republic of China, Russia, Iran, and North Korea) have an interest in replacing the United States as the primary global power, including through coercive and clandestine measures.
Current efforts are centered around National Security Presidential Memorandum 33 (NSPM-33). The memorandum was designed to deal with concerns related to a very specific problem: state-motivated concealment of ties with an adversarial military in activities at universities that were sponsored by the U.S. government. Obviously, concealment and fraud cannot and should not be rewarded in the U.S. system.
Over time, the bridge between concealment and espionage have proven difficult to establish in court, the research community writ large has (consequently) remained resistant to change, and the PRC has amplified the impact of U.S. research security measures and prosecutorial failures to drive a wedge between the government and members of the academic and Asian American communities.
It is prudent to recalibrate U.S. research security efforts to 1) maintain America’s ability to participate in global discovery by aligning research security efforts with associated risk of specific research activities and 2) create a clearer system for the identification of national security information (NSI) so as to enable protective measures, like the Espionage Act, to do their job. The status quo, which relies on the notion that information must remain in the open and be widely shared while somehow remaining out of reach of our adversaries, must be rationalized.
It is in the interests of the United States to appropriately protect information that needs to be protected while maintaining our participation in new discoveries to maintain our competitive advantage. Current efforts, which are focused on university faculty and partnerships, should be rebalanced to focus on risky technologies and ensuring that the source of most patented or sensitive technologies – the private sector – is adequately protected. Our current efforts are costly, excluding talented researchers with global connections from participating in the science and technology ecosystem and cutting the United States out of global discovery.
Challenge and Opportunity
There are a number of key challenges that a more effective research security regime will need to address:
NSPM-33 protects the government’s money from people, but it does not protect research or ideas from foreign appropriation
Ideas are the fundamental currency of technology competition. When a researcher or team of researchers have an idea, they frequently turn to their government for the financial backing necessary to nurture it. Under the NSPM-33 common forms requirement, the government’s evaluation of the proposal for funding includes an analysis of any conflicts of interest or commitment that individuals on the research team might have with an adversarial foreign government.
The research agency is expected to decline the funding opportunity if an applicant for funding has an ongoing or recent relationship with institutions that are backed by adversarial governments or military organizations (among other potential conflicts). In some circumstances, particularly when an individual or team intentionally misrepresents their relationships with adversarial governments, the federal agency may seek additional administrative remedies, including suspension and debarment. Similar processes are in place in government agencies that operate cooperative user facilities, primarily our national laboratories. Lying is bad, and it goes without saying that lying to conceal a relationship with an adversarial government is also bad.
Unfortunately, the story doesn’t end there. Because the idea is still wholly in the possession of the individual who applied for a federal grant, the individual or team is now free (and even incentivized) to seek alternative sources of funding. The fact that the government declined to support their work based on research security concerns as opposed to merit could validate the researcher’s belief that they have a meritorious idea worth pursuing elsewhere. A rare unscrupulous researcher could use that knowledge to determine the idea may be of interest to foreign militaries and actively look to adversarial governments for support. In effect, the government’s review of a person’s institutional affiliations, followed by a decision to exclude them from U.S. government funding, and subsequent visa revocations or revocations of permanent residency increases the probability of hostile foreign acquisition of talent and technology.
This conclusion is not based on speculation.
We’ve Been Here Before
History shows that strategic catastrophes follow when the government shuts people out of the American R&D ecosystem. In the 1950s, the United States detained and deported Qian Xuesen, co-founder of our Jet Propulsion Laboratory, based on fear that he supported the Chinese Communist Party. He was tapped to build the PRC’s ballistic missile and space programs, creating the strategic situation we find ourselves in today. Xuesen was so angry at the United States government that he later refused to take part in activities celebrating the normalization of relations. The United States made a similar error with Erdal Arıkan, the father of 5G wireless technology’s polar codes, albeit not out of concern for his affiliations. A lack of U.S. government support for long-term theoretical mathematics research led Arıkan to return from his positions in the U.S. to Turkey. Some years later, he was approached by Huawei. Huawei representatives quickly assimilated Arıkan’s work and became the world’s leading supplier of wireless technology. Subsequent government efforts to try to mitigate Huawei’s competitive advantage undoubtedly cost the U.S. taxpayer more than if we provided adequate or tailored funding for Arıkan’s career (and those of many other researchers) in an environment where it was more likely to be acquired by U.S. technology companies with less risk to the national security and the security of our international partners.
I have been briefed on other examples.
Funding Cuts Compound the Talent Crisis & Encourage Exodus
The Executive Branch’s massive funding cuts for research make the current situation even more problematic, creating compounding incentives encouraging talent to leave the country. Other countries have become better at attracting and retaining talent in recent years, weighing against the longstanding U.S. competitive advantage in talent attraction and retention. This has been particularly notable in fields like artificial intelligence, where particularly productive individuals have made significant contributions to the development of AI models like DeepSeek. The British-based talent intelligence firm Zeki Data points to the strengthening of markets for talent in Europe, the Persian Gulf, India, and China as additional reasons for a decline in U.S. AI talent attraction. Recent surveys suggest that as much as seventy five percent of the scientific community is looking for opportunities overseas. Notable recent departures from the United States due to green card denials, federal funding challenges, and federal layoffs include ChatGPT developer Kai Chen (moved to Canada), spaceflight safety expert Jonathan McDowell (moved to the UK), and carbon capture expert Yi Shouliang (moved to China). Neuroscientist Ardem Patapoutian was offered 20 years of funding in China in “any city, any university” after being cut off from NIH funding as part of the Trump Administration’s recent cuts, earning himself a mention during Marcia McNutt’s State of Science address at the National Academies.
Result: Innovation Increasing Elsewhere, Excluding Americans
When we assume that genius stems primarily from the U.S. system, and in particular from government funding, we are surprised when innovation happens somewhere else. The government’s principal method for controlling ideas and innovation only happens after a legal relationship between the researcher, their home institution, and the U.S. government is established. For advanced technology development, the overwhelming majority of which is funded by the private sector, we are principally reliant on export controls (which have a dismal history of successfully limiting knowledge and advanced technology transfer).
Science agencies aren’t adequately equipped to mitigate harm (and some NSI probably isn’t appropriately identified or protected)
For decades, National Security Decision Directive 189 (NSDD-189) was the primary governing document for research security measures. When it was released at the height of U.S.-Soviet tensions during the Reagan Administration, the academic community focused primarily on one line–”It is the policy of this Administration that, to the maximum extent possible, the products of fundamental research remain unrestricted.” NSDD-189 was meant to cover only a subset of research – fundamental research – defined in the document as “basic and applied research in science and engineering, the results of which ordinarily are published and shared broadly within the scientific community, as distinguished from proprietary research and from industrial development, design, production, and product utilization, the results of which ordinarily are restricted for proprietary or national security reasons.” On balance, the overwhelming majority of U.S. research performance and expenditure is not covered by NSDD-189, but rather developed by industry and the defense sector (which are covered later).
Understanding When Controls Must Be Used
Relatively little attention has been paid to NSDD-189’s second policy idea, which emphasizes that “where the national security requires control, the mechanism for control of information generated during federally-funded fundamental research in science, technology and engineering at colleges, universities and laboratories is classification.” Proper identification of potentially classifiable information and NSI is essential for the Justice Department and Intelligence Community, who wish to protect such research under national security statutes. The policy also acknowledges there may be alternative applicable statutes, like export control regimes, which have generally remained consistent with NSDD-189 (more on that later).
The classification system remains the most straightforward way to give our security community the tools they need to protect America’s critical national assets. This mirrors the findings of the 2019 JASON report on Fundamental Research Security, which argued that “Making the case, for classification reasons, that a new technology might be of national security value is far simpler than assessing its potential economic impact, even if economic security is equated in some way with national security.”
To fully appreciate what this means, one needs to understand when and why information should be classified. Classification is governed by Executive Order 13526 (E.O. 13526), which establishes the processes by which agencies establish national security classification protections. The lowest level of classification, which is termed “confidential,” is defined as information that “the unauthorized disclosure of which reasonably could be expected to cause damage to the national security that the original classification authority is able to identify or describe.” The bar for identifying and labeling “confidential” classified information is relatively low, and yet it is one that many science agencies lack the authority to implement.
Whether information should be treated as classified or unclassified is the responsibility of an Original Classification Authority (OCA). These authorities are a short list of senior government officials authorized by the President to review information and determine whether or not it is classified. Agencies that do not have representatives on the list are limited to being able to label information that is derived from other classified sources. To determine whether new information must be classified, individuals from other agencies must depend on determinations made by individuals granted OCA or delegated classification authorities.
Classification Constraints for NSF, NIH, others
The trouble begins when we realize that extramural funding agencies like the National Science Foundation (NSF) and National Institutes of Health (NIH) have no officials possessing OCA at the Top Secret (or even Secret) level, making officials in those agencies entirely dependent on members of the security community and White House to make such determinations. A broad lack of access to classified facilities limits the access that program managers have to relevant intelligence sources. Many NSF officials, for instance, need to travel offsite to engage with classified materials.
As a result, I have been told that classification reviews of new and novel information produced by these agencies almost never happens. If a researcher or program officer believes that the information that is produced in the course of research may damage national security and reports their concern to their funder in accordance with the terms of their grant (as is the case for NSF), the research agency lacks the authority to evaluate the information and defend their determination. Put another way, the research agency also lacks the authority to determine when edge-case information it produces should remain unclassified.
E.O. 13526 anticipates this and allows for the referral of information to original classification authorities at other agencies. Again, the need to refer elsewhere means that such reviews almost never happen, except for in those agencies which already possess the ability to identify and classify information (such as the Department of Defense or Department of Energy). Things get more absurd—one of the co-chairs in the Research Security Subcommittee of the National Science and Technology Council wasn’t able to read relevant research security-related intelligence products due to their reduced level of access. This had the effect of limiting the materials that could be discussed by the research security subcommittee.
Science Agencies Placed in a Bind
When an officer from a science agency or some other official makes the claim that the research they support could harm U.S. national security if it is improperly disclosed, the question “why wasn’t it classified” must immediately follow. A lack of original or derived classification authority means that the individual in question is not empowered to make the initial assessment of whether or not the information needs to be protected through the classification system. This effectively places them in conflict with the substance of E.O.13526 and unable to fully implement NSDD-189.
Defenders of the status quo would correctly point out the few examples of harm caused by the unauthorized disclosure of early stage research. While this is true, especially for agencies like the NSF or NIH, the inability to make a determination when paired with more recent Congressionally-directed emphasis on later-stage research (under organizations like the NSF Technology and Innovation Partnerships Directorate, ARPA-H, and other longer-standing programs involving gain of function research) increases the likelihood that later-stage research that could have national security implications will not be appropriately identified and protected. The expansion of research portfolios into applications must be accompanied by policies and a readiness to accept the resulting changes in responsibility.
The definition of basic and fundamental research has lost meaning over time and must be reestablished
During my time as Assistant Director for Research Security in the Office of Science and Technology Policy, security agencies would routinely point out that a fair number of basic research projects have fairly easily describable national security implications. I would agree with them and then take this one step further–a great deal of research that is categorized as basic research by science agencies fails to satisfy the statutory definition of basic research established for the Department of Defense. That definition is as follows:
Basic research is a systematic study directed toward greater knowledge or understanding of the fundamental aspects of phenomena and of observable facts without specific applications towards processes or products in mind. It includes all scientific study and experimentation directed toward increasing fundamental knowledge and understanding in those fields of the physical, engineering, environmental, and life sciences related to long-term national security needs. It is farsighted high payoff research that provides the basis for technological progress.
Similarly, the Federal Acquisition Regulations define the term as follows:
Basic research means that research directed toward increasing knowledge in science. The primary aim of basic research is a fuller knowledge or understanding of the subject under study, rather than any practical application of that knowledge.
The Export Administration Regulations (EAR) create two definitions to help establish what is considered fundamental research:
Fundamental research means research in science, engineering, or mathematics, the results of which ordinarily are published and shared broadly within the research community, and for which the researchers have not accepted restrictions for proprietary or national security reasons.
It is not considered fundamental research when there are restrictions placed on the outcome of the research or restrictions on methods used during the research. Proprietary research, industrial development, design, production, and product utilization the results of which are restricted and government funded research that specifically restricts the outcome for national security reasons are not considered fundamental research.
A fair number of defense research activities (including research supporting computing or hypersonics, as well as almost all “use-inspired research”) does not meet the statutory definition of basic research, but is often categorized as such despite the fact that the name of research or research field describes the applications or processes in mind. In the decades after NSDD-189, agencies have expanded on this definition in order to become inclusive of use-inspired research, muddying the waters.
For a final level-set, classified information and controlled unclassified information (CUI), cannot be defined as fundamental research in the eyes of the government as the act of controlling the information places the work outside the EAR’s fundamental research definition. This should be fairly straightforward given the fundamental incompatibility between the two definitions.
Applied Research Can Also Suffocate in a Closed System
Individuals who defend applied research activities being primarily handled in the open often point out that the overwhelming majority of these activities need to happen in the open in order to maintain technological competitiveness, pointing to the success and strength of open source technology platforms. I would agree and go one step farther: the vast majority of applied research would be worse off in a restricted environment, and there are many good reasons for why NSDD-189 included applied research in its definition of fundamental research. I also would point out that the strength of the EAR’s definition is also the acknowledgement that as soon as one feels the need to protect information from misappropriation, that information cannot and should not be managed in the open. It is incoherent to say that information needs to be in the open but also remain inaccessible by the Chinese government–the two notions are obviously mutually exclusive. To quote the JASON report on Fundamental Research Security, “The fundamental research exemption is based on the idea that the general nature of the knowledge produced in fundamental research cannot be controlled.”
Gold Standard Science and Industrial Diffusion Require Openness
While it is true that basic and fundamental research provide the basis for information that could eventually be used to support the development of sensitive and national security relevant technologies, expanding the “grey space” of risky information to include almost all basic research (as the Department of Energy and other departments are apparently now doing, as reported by some university officials) risks information paralysis. After all, literature review is an important first step in the scientific method, and shielding information from external eyes prevents research from being scrutinized in a way that results in the development of new questions or ideas, not to mention the entire system of scientific rigor. While it may be true that basic research may be used to develop security relevant technologies, it is not necessarily possible to entirely derive security relevant technologies from individual discoveries. Getting information into the hands of U.S. industrial players becomes much more difficult once it is restricted. This is especially for startups, small businesses, and smaller universities that cannot afford to sponsor vetting all of their employees at the level of “public trust”or higher in order to gain access to protected U.S. government-sponsored discoveries, creating consequences for American innovation, competition, and participation.
All of this is more difficult in a world where artificial intelligence can aggregate and synthesize information more rapidly than a team of graduate research assistants. Such risks must be accounted for. Still, it makes more sense to establish a strong governance framework around AI use cases rather than reordering how we govern the rest of society after every technological breakthrough.
Need to Limit the Grey Space
It is true that a great deal of research exists in a grey space where it needs to happen in the open in order to advance despite the fact that it may cause harm. Decisions to allow research that could cause harm to remain in the open should reflect the careful consideration of government program managers paired with unambiguous instructions to award recipients. It is in the capacity of the government (and not within the natural capabilities of our universities who have an incentive to seek the least restrictive framework possible) to determine and assess whether information might present a threat to national security and require that defensive measures be implemented, accordingly.
Muddying the waters around which research needs to be protected and which research does not has real world consequences for U.S. universities and non-governmental research organizations. When faced with ambiguity about whether or not they should be engaged in a particular research activity, university administrators frequently take the most cautious approach possible to protect themselves from potential liability and reputational damage. The talent, on the other hand, retains their ability to choose to do their work elsewhere if an environment becomes too restrictive.
Securitization has significant costs (including to American leadership)
Later in his life, Richard Feynman recounted a story about the impact of compartmentalization on the Manhattan Project. Uranium-235 separation happened at Oak Ridge National Laboratory while the research underpinning that production took place at Los Alamos. Because the researchers at Oak Ridge didn’t have the fundamental understanding of nuclear physics necessary to troubleshoot their work, Uranium-235 production ran into numerous headwinds until Feynman and others managed to convince the government of the necessity of sharing knowledge with the Oak Ridge team. Once the knowledge was shared, production went back on track. Failure to share the information with other scientists risked a major accident at Oak Ridge, not to mention an end to the Manhattan Project. In today’s environment, where incentives are structured in such a way that critical decisionmakers frequently sit on research security-related decisions rather than subjecting themselves to internal or Congressional scrutiny, I am less confident that someone like Feynman would reach a similar outcome.
Measuring Direct and Indirect Costs
It is relatively easy to measure the financial costs associated with research security measures. It is much more difficult to evaluate their effectiveness. The fact that there have been few publicized significant examples of national security harm resulting from the sharing of government-sponsored basic research makes having a serious discussion about the efficacy of our existing research security measures outside of classified environments almost impossible.
Costs include increased administrative burden, support staff, defensive cyber systems, facility access controls, and more. These costs cannot be easily accounted for in an individual grant’s direct costs, so we can safely assume that decreases in support for indirect costs under the current administration will result in the cost of CHIPS and Science-mandated research security programs placing additional pressure on other scientific activities. Research security compliance costs are overwhelmingly treated by the government as mandatory expenditures, while (confoundingly) the cost of operating a world-class laboratory is not (except for in instances where a grant is explicitly for the development and maintenance of advanced scientific infrastructure).
Costs to American Competitiveness
While we can also measure the economic impact of fewer students and scholars in the United States, it is more difficult to measure the cost of the United States being absent from international opportunities. Proposed legislation like the SAFE Research Act, while intended to prevent U.S. government funding from supporting research involving adversarial nations, effectively could result in U.S. researchers being forced to abandon high-potential discoveries produced by large and diverse international research consortia if researchers from adversarial countries are part of the team. This would give the PRC veto power over the involvement of the U.S. government-supported individuals in international research consortia–an alarming consequence, especially when one considers the Australian Science Policy Institute’s recent finding that Chinese research institutions dominate 57 out of 64 critical technology fields and the fact that some international projects require the work of thousands of scientists from dozens of countries. In fields like space research and development, the PRC’s domestic talent base is sufficient to maintain competitiveness while they actively test capabilities where the United States is years away from deployment (and might beat the United States back to the moon).
The absence of U.S. researchers in such consortia does not mean that the research does not happen–it merely means that the consortium needs to find other partners who are able to do the work. The fact that the infrastructure supporting such discoveries is frequently located exclusively in Europe and Asia (thanks in part to decades of U.S. underinvestment in research and development infrastructure) means that there is additional incentive for American researchers to travel and work abroad in order to maintain access to unique capabilities located in adversarial countries to advance their careers and test new technologies.
This problem is particularly acute in fusion energy research and development, where U.S. companies maintain a technological advantage but lack the domestic extreme radiation environment testing infrastructure necessary to develop critical components necessary for long term operation (the inner walls of the reactor, or blanket).
A protect-oriented research security framework makes sense if the United States is safely in the lead and competitors are decades away from catching up. But in a more competitive environment, when other countries become more productive and competitive, decisions that force the United States to abandon collaborative efforts are much more consequential. Given the relative size of the U.S. population with that of our competitors, this mitigates the ability of the United States to unilaterally define global rules governing the conduct of research.
Taken together with my previous point about the need to re-establish the definition of basic and fundamental research as a bright line test and giving science agencies greater ability to control information, I’m placing an awful lot of responsibility on the backs of federal research managers. Having spent most of my professional life in the civil service as a clearance-holder, I would argue that managing such weighty decisions is exactly what federal employees are paid to do. In the same talk mentioned earlier, Feynman described the admiration he had for the speed with which members of the security establishment could make decisions upon which the fate of the nation rests. If we abandon the flexibility of officers to give informed answers to our research apparatus promptly, we do so at our own peril.
We know dangerously little about what is happening in Chinese laboratories, and fear is getting in the way of dealing with our knowledge gaps and urgent security challenges
As a former CIA colleague recently briefed Congressional staff, it is very difficult for trained intelligence officers from both the United States and the PRC to successfully infiltrate the academic environment due to the level of academic training required and fluency in domain-specific technical terms and practice. When the United States occupies a position of preeminence, and the primary sources of technical knowledge originate in American laboratories, then it makes sense to limit partnerships with individuals and organizations who have competing interests. Unfortunately, the United States cannot count on itself being in the lead in many scientific domains any longer. Our ability to avoid technological surprise depends, in part, on knowing what’s going on in Chinese labs.
The research security push over the past decade makes this much more difficult. The growing absence of routine technical interactions with Chinese counterparts means that our scientists and engineers have limited insight into the PRC’s work. The emphasis of research security requirements aims to restrict interactions with the PRC’s Seven Sons of National Defense, which also happen to be the PRC’s top performers in science and technology discovery. Without knowledge of what is happening in these institutions or the ability of our scientific community to ground-truth intelligence assessments (which are frequently compiled by non-experts), we increase the probability that we will not have the immediate knowledge necessary to replicate such discoveries or worse, fall victim to technological surprise.
Degraded Capacity Creates Larger Competitiveness Risks
The challenge is compounded when we consider the degradation of our own technical capacity. In fields like nuclear fusion and radio astronomy, the United States simply does not have the laboratory capabilities that are necessary for our scientists to remain at the cutting edge. In 2023, the Fusion Energy Science Advisory Committee found that “rapid progress toward commercial fusion power will likely rely in part on research at existing or near-term international facilities that provide capabilities presently unavailable in the U.S., such as long-pulse magnetic confinement” – an area where the PRC has invested billions of dollars in recent years. Decoupling and fear of information transfer also has limited our ability to communicate with the PRC in areas like spaceflight safety, where we generate dozens of conjunction warnings every day involving potential near-misses with Chinese satellites. The consequences of non-communication or inefficient communication with PRC satellite operators resulting from a collision cascade includes the loss of access to particular orbits, significant damage to U.S. and allied commercial and security assets, and potentially unlimited liabilities for the United States under the Outer Space Treaty. Such a disaster would have dire consequences for our economy and space-dependent national security establishment.
This creates a Catch-22 for American scientists and engineers who want to see the United States advance in the field or see their fields advance. On the one hand, testing new technologies on foreign platforms creates substantial risk of foreign appropriation, similar to the risks experienced by American companies operating in China since normalization. On the other, the lack of access to similar domestic or allied capabilities freezes the ability of U.S. commercial interests to keep pace with foreign competitors.
Increased Departures, Stunted Discovery Process
The U.S. government, likewise, cannot count on individuals who have devoted their lives to a particular field of inquiry to wait for the government to invest in these capabilities in the next five to ten years (an overly optimistic projection for the budget process, alone). History shows us that brain drain (or loss) in such situations is inevitable. America’s ability to remain competitive will increasingly depend in some part on our access to PRC-derived knowledge sources absent new and significant U.S. government investment. A research security regime that unnecessarily restricts U.S. collaborations based on institutional affiliation or connections, rather than domains where the risk of loss exceeds potential benefits, is more likely than not to limit our insight into new knowledge early in the discovery process, and throttle U.S. technological development in a way that will make it more difficult to compete in the future.
Research integrity has become securitized, confusing the ways in which we approach domestic challenges
Research integrity and research security are interlinked, and it is absolutely true that when contracts developed by a foreign government encourage researchers to lie on federal grant applications that is a major cause for concern and demands a national-level response. But no matter how egregious such behavior is, and no matter how proximate violations of the False Claims Act are to violations of the Espionage Act, they are different parts of the criminal code. If the government wishes to bring a case before the courts where an individual has both lied to the government on official documents and provided NSI to a foreign government, it is in the power of a prosecutor to do so and seek appropriate penalties.
On the domestic side, organizations such as Retraction Watch have justifiably brought attention to research misconduct over the past ten years. There are significant examples of senior researchers, including 13 retractions from a Nobel laureate, of having engaged in alleged violations of research integrity. Many of these cases involve the knowing manipulation or falsification of data. I would argue that such cases do more to damage the integrity of the U.S. research ecosystem than individuals omitting information about their affiliations in grant applications, primarily because the publication of falsehoods into the scientific corpus, exacerbating the “replication crisis” in biology, psychology, and behavioral economics, and poisoning the scientific establishment’s credibility.
In several recent domestic misconduct cases, the researchers in question were able to return to their laboratories, continue their day jobs, and even found unicorn technology startups. On the other hand, undisclosed affiliations, patents, or grant support have resulted in the termination of 119 scientists (almost half) investigated by the National Institutes of Health as part of their foreign interference efforts. Some researchers have been denied access to funding despite no credible link to foreign talent programs or other similarly concerning affiliations. A reasonable observer would be left to wonder why an undisclosed patent or grant support is a greater violation than the intentional falsification of the results of their research. To the observer the answer may seem obvious–in one instance, we have a nexus with a foreign government; in the other instance, we don’t.
As former Assistant Attorney General Matt Olsen noted when the Department of Justice drew down the China Initiative in 2022, “by grouping cases under the China Initiative rubric, we helped give rise to a harmful perception that the department applies a lower standard to investigate and prosecute criminal conduct related to that country or that we in some way view people with racial, ethnic or familial ties to China differently.” The same must be true of administrative actions related to academic misconduct, especially as countersuits undermine the government’s earlier claims.
Proper NSI Identification Removes Ambiguity, Releases Burden
This is why it is important that national security-sensitive research be clearly identified as NSI through the classification system. Once information has been appropriately classified, then who gets to handle the information becomes just as important as how the information is handled. Individuals who handle classified information are expected to report their contacts, affiliations with foreign governments, and all other information that is necessary to maintain the public trust as a matter of national security. Violations related to improper handling may be appropriately elevated under the Espionage Act and successfully prosecuted in court. Our current attempts to substitute judicial action with administrative penalties creates a culture of fear and suspicion in our university establishment, rhyming with some of the darkest periods in the history of our nation’s research and development enterprise. The culture of fear, compounded with a lack of clarity about what needs to be protected, dramatically increases the probability that universities will improperly exclude individuals from certain racial or ethnic backgrounds on activities that have little relevance to national security.
Plan of Action
Recommendation 1. Congress should, with the support of the Administration, reinvest in American research and innovation, including in foreign talent attraction.
The United States is no longer in a position where American technological superiority is assured; the fact that the PRC is leading in publications in many critical and emerging technology fields, and in the deployment of advanced technologies in certain domains, should be of immediate cause for concern. Likewise, anticipated declines in foreign talent enrollment in U.S. universities and shrinking PhD programs at major U.S. universities in response to federal policy actions should be of significant cause for concern. In 2024, the Defense Department reported that “unfunded research, development, test, and evaluation (RDT&E) infrastructure requirements were shown to have grown significantly since annual reporting began in 2018, putting the military at risk of losing its technological superiority.”
Funding also provides the government with the leverage needed to protect American interests. The Department of Defense’s use of contracts to protect U.S. security interests is already common practice, most famously recently used to ensure that SpaceX’s Starlink would not cut service in Ukraine. Similarly, the government cannot classify, restrict, or otherwise currently control information that is produced outside the federal research ecosystem. Even if it is possible that an idea may make it back to a foreign government or adversarial military, it is far better for the United States to have participated in the development of that idea (and have the chance to exploit relevant intellectual property) than it is to surrender the innovation, in total, to a foreign power, robbing us of the chance to compete. Sometimes, in order to control the dissemination of an idea, the government must invest in it, and that means that the government should actively seek to invest in projects in order to participate in the discovery and gain access to its benefits, even when our partners might be less than ideal or when there is risk that the information could also be used by a hostile foreign power.
The government can and should create terms in its funding mechanisms that enable it to mitigate risk where necessary, as well as giving it graduated negotiating flexibility in circumstances where we may require enhanced measures (like encouraging researchers to sever ties with adversarial entities like the PRC). These should not be mandatory, as is the case with the SAFE Research Act, which effectively gives the PRC the power to veto U.S. participation in large multinational consortia if individual PRC researchers become affiliated with an effort, thereby reducing the ability of the United States to set terms for increasingly multilateral scientific activities.
Recommendation 2. The Office of Management and Budget (OMB) should require rigorous and independent cost-benefit analysis of existing research security efforts before approving new research security-related regulations and requirements as part of the Paperwork Reduction Act review process. Congress should exercise similar care before imposing similar requirements on academic institutions given the significant costs associated with research security programs.
The use of grants to impose research security requirements that alter an organization’s behavior at the institutional level are, in effect, de-facto regulatory action with significant economic impact (as defined in Executive Order 12866). As attesting that an institution has a research security program that meets the requirements of a granting agency requires significant economic expenditure across many academic institutions, new research security requirements should be subject to the same cost-benefit analysis as other significant regulatory actions. Paired with declining support for indirect costs and limited public evidence that disclosures of federally-supported basic research has harmed U.S. national security interests, there is a financial imperative to ensure that research security programs are aligned with actual instances of observed harm to national security and the unintended or illicit transfer of national security information (NSI).
Consistent with recommendations from the recent National Academies report on simplifying research regulations and policies, existing research security efforts should be recalibrated to mitigate risk of loss as opposed to blanket bans on interactions as a result of institutional affiliation. Efforts that result in reduced American participation in global discovery around critical technologies directly cut against our country’s technological competitiveness and should be retired and rescinded, as the most likely outcome of such measures is not the protection of information, but rather the inability of the United States to develop and deploy new and novel capabilities.
What could the costs for expanded research security programs look like, especially given that some agencies are allegedly treating basic research activities as CUI? As noted in the JASON report on Safeguarding the Research Enterprise:
“The supporting apparatus for access controls would impose significant cost on the conduct of research and reduce research funding efficiency. JASON received from NSF cost estimates for what the University of Oklahoma has spent to support such work, for example. A warehouse-type building for CUI experiments was estimated to have cost $2M, and a new office building with access control adequate for classified work cost $7M. Building construction costs are only about 10–20% of their life-cycle ownership costs, translating to roughly $1–2M per year for both buildings. Required security and compliance staff add cost of four full-time equivalent personnel, equating to another $1M per year. Thus, a medium to large ($1–3M/year) research program might incur security costs around $1–3M per year above the baseline research cost, roughly doubling the cost of carrying out that research. This would constitute a serious loss of research efficiency. Slowing research by half could easily allow countries like the People’s Republic of China (PRC) to pull ahead in strategic fundamental research areas.”
Strangling our research enterprise is an unacceptable outcome and must be avoided. Likewise, expanding research security reviews within agencies would likely require significant expansion of agency personnel available to conduct rigorous national security assessments and cut against the availability of funds to do actual science. Such a tax on research funding is an additional cost to American competitiveness, reduces the number of grants made to institutions, and serves to further exclude institutions in EPSCoR jurisdictions, HBCUs, and other MSIs, emergent research institutions, and nonprofit laboratories that already face significant resource constraints.
In light of preliminary assessments of cost to the research enterprise, the Government Accountability Office (GAO, which is within the legislative branch) may wish to also assess the effectiveness of research security programs compared with their cost of implementation, and in particular the impact on programs that do not have a clear NSI nexus.
The Administration and Congress must ask if they are prepared to significantly increase funding for research programs in order to deal with such significant cost inflation due to mandatory research security program requirements. The situation will only become more dire if the government caps indirect costs at 15 percent, as several science agencies recently attempted to impose, which would place research security costs in even greater conflict with the fundamental resources necessary for an institution to conduct research. Such security measures are meaningless if the research institution lacks the staffing, facilities, or administrative support necessary to conduct cutting-edge research. Care should be taken to ensure that federal policy actions attempting to reign in administrative costs do not cut against our ability to operate world-class research institutions.
Recommendation 3. Congress should figure out how best to handle research security concerns that originate from research supported by non-governmental entities, and empower industry consortia to take measures to secure their sensitive intellectual property. Incentivizing participation through contracts, cost deferrals, or reduction of administrative burden for protective measures can help.
NSDD-189 warns that “as the emerging government-university-industry partnership in research activities continues to grow, a more significant problem may well develop.” Contemporaries involved in the drafting of NSDD-189 have told me that the growing role of the private sector in funding university research was of concern to Reagan’s OSTP, and that as industry became a more prominent funder, then the government would have limited controls to prevent the transfer of industry-derived information to foreign powers. Given the increased presence of commercial research laboratories, and more recently focused research organizations, the emphasis of research security efforts on federal funding for universities is inconsistent with the balance of U.S. research performance.
The majority of research considered proprietary or sensitive (as well as the overwhelming majority of all U.S. research) is produced or supported by American companies. As a matter of policy, most universities will not accept controls on the research which they produce, appropriately intending for it to enter into the public domain where it can be more rapidly developed and used for practical benefit. Academia’s role as a producer and broad distributor of knowledge is fundamental to its role in society and must be protected. Rather than suggesting that most university research is sensitive, when its primary purpose is to be shared, Congress should be willing to enact enforcement mechanisms that strengthen the ability of the government to protect research relevant to defense and industry.
On this point there are no easy answers. The danger, of course, is that such measures will inherently restrict the competitiveness of American businesses and their ability to participate in international markets. Strengthening CFIUS can hinder the ability of American companies to secure sources of investment and invite foreign retaliation. Expanding our use of export controls will create incentives for foreign governments to diversify away from American suppliers and limit the ability of our companies to shape global supply chains. Restricting foreign countries’ access to U.S. technology will instead create new dependencies on foreign technology sources, creating a new system of incentives that limit the ability of the United States to set global standards, and force countries to make concessions to the PRC in order to maintain access to their resources and technology. Our efforts to limit the PRC’s access to semiconductors has already convinced the European Union of the need to become less dependent on the United States in other technology areas. The difficulty the United States has had in managing the spread of technology from Huawei, Bytedance, and BYD should be instructive, as should growing challenges to U.S. influence in multilateral fora.
The most straightforward way to control information produced outside the government is by getting knowledge producers on contract and creating a system of incentives for companies to do so. Doing so has two purposes. First, it allows the government to create a financial incentive for companies to participate in enhanced security measures. Second, it creates a legal relationship where companies can be required to implement enhanced security measures without significant sacrifices to their bottom line. This could be through offering to defer costs associated with security clearances and technology measures needed to provide enhanced security and IP protection. Legal relationships are more effective than education campaigns, given that some producers of high value technology don’t seek government protection given the significant financial and administrative costs. Still, some firms may refuse to take government grants or contracts to preserve their organizational flexibility. This is a feature of American capitalism, not a bug.
Managing these tradeoffs is fundamental to addressing our research security challenges. If we want to get serious about addressing research security, our greatest efforts need to be directed to research that is higher in the value chain with national security consequences. Most of that research doesn’t happen in universities. Policymakers must be prepared to accept the inherent tradeoffs that come with decisions to place restrictions on the American research enterprise.
Recommendation 4. The Executive Branch should grant Original Classification Authority to the heads of extramural granting agencies and more strictly apply the federally-recognized definitions of basic and fundamental research.
As federal extramural funding agencies move toward more technology-relevant solutions and later-stage technology development and deployment, the ability of agencies to defend decisions to keep research in the open environment will become much more acute. While the risks of overclassification are real, the risks are not greater than in fields like diplomatic engagement or global development (both by definition require continuous engagement with foreign partners, especially with those who wish to challenge American interests). The Department of Defense and Intelligence Advanced Research Projects Activity (IARPA) experience and history of prudent and limited classification determinations should provide some relief.
This will not be cheap, and agencies will need appropriate resourcing in terms of personnel and capital to manage the workload and specialized technology facilities (Sensitive Compartmented Information Facilities, or SCIFs, and secure work areas) to manage classified information.
More strictly applying the federal definition of basic research to research that is “without specific applications or processes in mind” should help agencies separate activities that have clear industrial, commercial, or defense-related applications from research that is truly foundational. The task of establishing a “bright line” between applications with national security potential is likely to be significant, but it is likely to be far less costly than passing that cost onto thousands of laboratories around the country to make their own risk management calculations based on less complete information. NSF has created the SECURE Center to mitigate this, but again, the solicitation for the Center notes that the Center does not conduct investigations, hold or manage classified information, or assume liability for the consequences of its products.
Agencies will also need to deal with the fact that many universities and other non-governmental research organizations frequently do not accept funding that comes with classification, CUI, or other burdensome requirements. In such instances agencies will need to weigh the value of the research in question and balance that with the risk of foreign appropriation. In instances where the risk of foreign appropriation is significant, the value of the research is high, and the consequences of the United States not participating in discovery are anticipated to be significant, agencies should be willing to provide supplemental funding to enable the research activity to take place and to devote an appropriate level of oversight to ensure that U.S. involvement in the activity is appropriately managed and consistent with overarching federal interests.
The government might also wish to implement alternative funding measures when there is an interest in participating in discovery while excluding the participation of non-aligned governments. Using the contract or collaborative agreement systems are probably more appropriate than grants in such circumstances, allowing the government to more expressly dictate terms for collaboration. The government should be willing to use financial leverage to do this, including through funding for large international research consortia, to counter the efforts of adversarial governments while maintaining the ability of the United States to participate in overseas discovery processes.
Yes, this will result in a culture change in some of our premier research-supporting agencies. I would argue that culture change became necessary immediately after the creation of the NSF TIP Directorate, ARPA-H, and similar organizations. If we expect these agencies to engage more directly in critical and emerging technology development, especially around technologies which are relevant to great power competition, then increased scrutiny of these and similar programs is necessary to protect our critical national assets.
Recommendation 5. The Executive Branch should move all research integrity efforts under the rubric of Gold Standard Science along with other issues related to academic misconduct.
This is not a lengthy recommendation; the integrity of our efforts to preserve and protect the integrity of the research enterprise should be managed under a single umbrella where penalties for unethical conduct can be calibrated to the severity of the misconduct. This is essential for maintaining buy-in within the research community around both research security and research integrity measures. Invoking Justice Holmes, if we take the view of an unscrupulous researcher (whom we shall find does not care two straws for ethical conduct in the sciences but does want to know what will result in a loss of tenure) then it is reasonable for them to assume that the government’s ethical framework for science is mediated primarily by our relationship with Chinese research institutions as opposed to Gold Standard Science. It is in the interest of the science and technology ecosystem to correct that notion at the earliest possible opportunity (and presumably to also clarify when the government will charge them with espionage).
Conclusion
It is highly probable that this administration and Congress will seek to implement additional measures related to research security in the near term. Current and proposed frameworks incorrectly assume persistent U.S. supremacy in science and technology and that new ideas are a product of government innovation. As the PRC deploys new capabilities that have not been demonstrated by U.S. government or commercial actors, the posture of the United States toward our research security apparatus must change to match the strategic moment. Measures that isolate the United States from discovery should be retired in favor of new measures that selectively identify and protect critical knowledge vital to maintaining national security. For the sake of our security and future technological leadership, we must recognize that innovation comes from the work of teams of individuals who are motivated to change the world, and accept that America is less secure when that change happens somewhere else.