An Innovation Agenda for Addiction: Breakthrough Medicines That Scale
The federal government should expand the FDA’s priority review voucher program (PRV) and provide market exclusivity advantages to encourage the development of medications for addiction.
Taken together, substance use disorders (alcohol, cigarettes, and other drugs) cause more deaths in the U.S. every year than cancer or heart disease and cause devastating downstream social harms. Despite this, only 3% of eligible patients received substance use disorder (SUD) medication, a result of low uptake and efficacy of existing medications and a lack of options for patients addicted to stimulants. This is due to a near-total absence of pharmaceutical research and development activity. To make population level impact to reduce harms from opioids, methamphetamine, cocaine, alcohol, and cigarettes, we must address the broken market dynamics in addiction medicine.
The PRV program should be expanded to cover opioid use disorder, alcohol use disorder, stimulant use disorder, and smoking. In addition, drugs that are approved for these SUD indications should have extended exclusivity and sponsors that develop these medications should receive vouchers to extend exclusivity for other medications.
Challenge and Opportunity
Addiction policy efforts on both the left and the right have struggled. Despite substantial progress reducing smoking, 29 million Americans still smoke cigarettes and feel unable to quit and 480,000 Americans die each year from smoking. While overdose deaths from opioids, cocaine, and methamphetamine have fallen slightly from their peak in 2022, they are still near record highs, three times higher than 20 years ago. Alcohol deaths per capita have doubled since 1999.
Roughly 60% of all crimes and 65% of violent crimes are related to drugs or alcohol; and the opioid crisis alone costs the United States $1.5 trillion a year. Progress in reducing addiction is held back because people with a substance use disorder take medication. This low uptake has multiple causes: in opiate use disorder, uptake is persistently low despite recent relaxations of prescription rules, with patients reporting a variety of reasons for refusal; treatments for alcohol use disorder have modest effects; and there are no approved treatments for stimulant use disorder. Only three percent take SUD medications, as shown in figure 1 below [link to image]. In brief, only 2% of those suffering alcohol use disorder, 13% of those with opiate use disorder, 2% of smokers, and approximately 0% of illicit stimulant users are receiving medication, giving a weighted average of about 3%.
There has been rapid innovation in the illicit market as synthetic opioids and expanded meth production have lowered price and increased strength and availability. Meanwhile, there has been virtually no innovation in medicines to prevent and treat addiction. The last significant FDA approval for opioid use disorder was buprenorphine in 2002; progress since then has been minimal, with new formulations or dosing of old medications. For alcohol use disorder, the most recent was acamprosate in 2004 (and it is rarely prescribed due to limited efficacy and three times a day dosing).
None of the ten largest pharmaceutical companies have active addiction medicine programs or drug candidates, and the pharmaceutical industry as a whole has only pursued minimal drug development. According to the trade association BIO, “Venture investment into companies with novel addiction drug programs over the last 10 years is estimated at $130M, 270 times less than oncology.”
There are promising addiction drug candidates being studied by academics but without industry support they will never become medicines. If pharmaceutical companies spent just 10% of what they spend on obesity therapies, we might quickly make progress.
For example, GLP-1 medicines like Ozempic and Mounjaro have strong anti-addictive effects across substances. Randomized trials and real-world patient health record studies show dramatic drops in consumption of drugs and alcohol for patients taking a GLP-1. Many addiction scientists now consider these compounds to be the biggest breakthrough in decades. However, Novo Nordisk and Eli Lilly, who own the drugs currently in the market, do not plan to run phase 3 addiction trials on them, due to fear of adverse events in substance use disorder populations. The result is that a huge medical opportunity is stuck in limbo indefinitely. Fortunately, Lilly has recently signaled that they will run trials on related compounds, but remain years from approval.
Conversations with industry leaders make clear that large pharmas avoid SUD indications for several reasons. First, their upside appears limited, since current SUD medications have modest sales. Second, like other psychiatric disorders, the problem is challenging given the range and complexity of neurological targets and the logistical challenges of recruiting people with substance use disorder as participants. Finally, companies face downside reputational and regulatory risk if participants, who face high baseline rates of death from overdose regardless, were to die in trials. In the case of Ozempic and Mounjaro, sponsors face an obstacle some have termed the “problem of new uses” – clinical trials of an already lucrative drug for a new indication carry downside risk if new side effects or adverse events are reported.
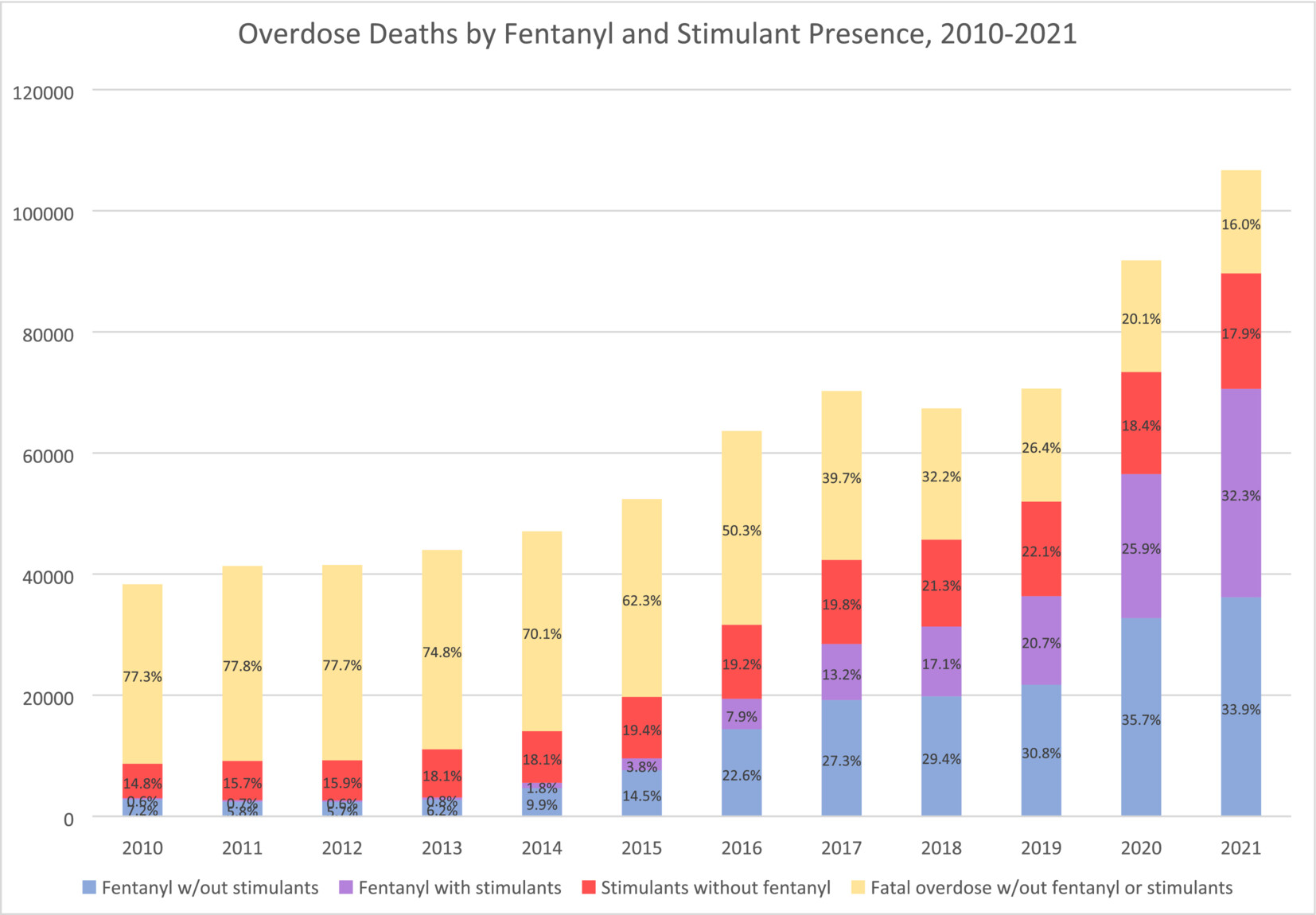
Image from Charting the fourth wave, based on CDC data
Plan of Action
Market Shaping Interventions
Recommendation 1. Expand the FDA priority review voucher (PRV) program to include addiction medicine indications.
The FDA priority review voucher (PRV) program incentivizes development of drugs for rare pediatric and infectious diseases by rewarding companies who get drugs approved with a transferable voucher that accelerates FDA approval. These vouchers are currently selling for an average of $100M. The PRV program doesn’t cost the government any money but it makes drug development in the designated categories much more lucrative. The PRV program has proven very successful, leading to a surge in approvals of medications.
As a neglected market with urgent unmet medical and public health needs, and which also promises to benefit the broader public by reducing the negative externalities of addiction, addiction medicine is a perfect fit for the PRV program. Doing so could transform the broken market dynamics of the field. The advantage of the PRV program is that it does not require substantial new congressional appropriations, though it will require Congress giving the FDA authority to expand the PRV program, as it has done previously to add other disease areas.
Recommendation 2. Extend exclusivity for addiction medicines and incentivize pursuit of new indications
Market exclusivity is a primary driver of pharmaceutical industry revenue. Extending exclusivities would have a very large effect on industry behavior and is needed to create sufficient incentives. The duration of exclusivity for alcohol use disorder, opioid use disorder, stimulant use disorder, and smoking cessation indications should be extended along with other incentives.
- Addiction medicine indications should receive an additional two years of exclusivity for biologics and three years for small molecules.
- Companies that achieve an indication for a substance use disorder for a medication that represents a significant advance would receive an exclusivity voucher that can be transferred to another medication. For 2nd, 3rd, and 4th SUD indications with the same compound, companies would be granted a shorter duration exclusivity voucher. Durations would be tiered, as described in this proposal from Duke, to balance public interest and reward levels.
- FDA should provide increased consideration for addiction medicines for breakthrough, fast track, and priority review designations, as well as accelerate meeting schedules, all of which substantially reduce development expenses.
For precedent, there are already a number of FDA programs that extend medication exclusivity, including ‘orphan drug exclusivity’ and the qualified infectious disease product (QIDP) program. Like rare diseases and antibiotics, addiction is a market that requires incentives to function effectively. In addition, successful treatments, given the negative externalities of addiction, have public benefit beyond the direct medical impact, and deserve additional public incentives.
Recommendation 3. Modernize FDA Standards of Efficacy for Substance Use Disorder Trials
A significant barrier to pharmaceutical innovation in SUDs is outdated or unpredictable efficacy standards sometimes set by the FDA for clinical trials. Efficacy expectations for substance use disorder indications are often rooted in abstinence-only and other binary measure orientations that the scientific and medical community has moved past when evaluating substance use disorder harms.
This article in the American Journal of Drug and Alcohol Abuse demonstrates that binary outcome measures like ‘number of heavy drinking days’ (NHDD) can underestimate the efficacy of treatments. This recent report from NIAAA on alcohol trial endpoints recommends a shift away from abstinence-based endpoints and towards more meaningful consumption-based endpoints. This approach should be adopted by the FDA for all SUD treatments, not just alcohol.
There are some indications that the FDA has begun modernizing their approach. This recent paper from NIH and FDA on smoking cessation therapies provides updated guidance that moves in the right direction.
More broadly, the FDA should work to adopt endpoints and standards of efficacy that mirror standards in other disease areas. This shift is best achieved through new guidance or statements issued by the FDA, which would offer positive assurance to pharmaceutical companies that they have achievable paths to approval. Predictability throughout the medication development life cycle is absolutely essential for companies considering investment.
Congress should include statements in upcoming appropriations and authorizations that state:
- The FDA should adopt non-binary standards of efficacy for addiction treatments that are aligned with standards for other common disorders and the FDA shall, within 12 months, report on the standards employed for substance use disorder relative to other prevalent chronic conditions and report steps to eliminate disparities in evidentiary standards and issue new guidance on the subject.
- The FDA should publish clear guidance on endpoints across SUDs to support planning among pharmaceutical companies considering work in this field.
Conclusion
Sustained focus and investment in diabetes and heart disease treatments has enabled medical breakthroughs. Addiction medicine, by contrast, has been largely stagnant for decades. Stimulating private-sector interest in addiction medicine through regulatory and exclusivity incentives, as well as modernized efficacy standards, is essential for disrupting the status quo. Breakthroughs in addiction medicine could save hundreds of thousands of lives in the US and provide long-term relief for one of our most intractable social problems. Given the negative externalities of addiction, this would also have enormous benefits for society at large, reducing crime and intergenerational trauma and saving money on social services and law enforcement.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
Per author conversations with industry leaders, private sector interest in SUD medication development is limited for the following reasons:
- The upside of pursuing SUD indications appears limited, since current SUD medications, which are generally targeted for specific substances, have modest sales.
- Even with preliminary evidence that GLP-1 drugs may be efficacious for some SUD indications (e.g, alcohol, opiates, and tobacco), companies are reluctant to pursue label expansion for SUD. As described previously, with already lucrative drugs, companies face a downside risk (termed the “problem of new uses”) from running large clinical trials, and possibly uncovering new side effects or incurring random adverse events which could harm reputation and existing markets.
- In the specific case of SUD, this downside risk might be especially large, since people with substance use disorder have high baseline rates of overdose and death.
Moreover, there is an argument that a treatment for SUD is a public good, to the degree that it ameliorates the negative externalities of addiction – increasing the case for more public-sector incentives for SUD treatment. The end result is that medical treatments for SUD are stuck in an indefinite limbo, with private-sector interest in SUD, as documented previously, being very low.
The current lack of effective and widely used SUD medications is disheartening, but this is in the context of private sector disinterest and scant funding. Even modest successes in SUD treatment have the potential to kickstart an innovation loop, akin to the rush of biotech companies hastening to enter the obesity treatment field. Prior to the success of the GLP-1 drugs, obesity treatment had been moribund, and viewed pessimistically in light of drugs that had limited efficacy or had been withdrawn for side effects like suicidality or cardiovascular issues.
An SUD success like GLP-1 for obesity has the potential to kindle a similar rush of interest; the challenge is the initiation of that cascade. Given the very low levels of investment in SUD treatments, there is potential low-hanging fruit that, given sufficient funding, could be trialed and deployed.
There has been rapid innovation in the field of addiction, but it’s been happening on the wrong side: addiction-inducing technologies are becoming more powerful, while SUD treatments have largely stagnated. This innovation is most evident in synthetic opioids and methamphetamine.
Compared to heroin, fentanyl is about 25x stronger (on a per-weight basis), and hence, much easier to smuggle. As the Commission on Combating Synthetic Opioid Trafficking put it:
Single-digit metric tonnage of pure fentanyl is not a large amount and could easily fit into a shipping container or a truck trailer, which seriously challenges interdiction…Perhaps as much as 5 MT [metric tons] of pure fentanyl would be needed to satisfy the entire annual U.S. consumption for illegally supplied opioids.
Moreover, as a recent Scientific American article documented, innovations in fentanyl production, including the use of safer precursors and methods that don’t require sophisticated equipment, mean that fentanyl production is now decentralized, and resistant to attempts by law enforcement to shut it down.
As fentanyl has come to dominate the opioid supply over the past 10 years, overdose deaths have risen dramatically. New synthetic opioids and non-opioids like xylazine are also becoming common.
At the same time, due to advances in production techniques in Mexico, methamphetamine production has skyrocketed in recent decades while purity has improved. Worst of all, unlike heroin, fentanyl is easily combined with meth and cocaine in pills and powder.
The DEA has highlighted the presence of “super labs” in Mexico capable of producing hundreds of pounds of meth per batch.
Together, these three innovations (fentanyl, cheap meth, and new combinations) have led to a 400% increase in overdose deaths in the past 20 years. Without equally powerful innovations to reduce addiction rates, we will never make long-term and sustainable progress.
Ready for the Next Threat: Creating a Commercial Public Health Emergency Payment System
There are many examples of groundbreaking success in the development of life-saving vaccines, diagnostics, and therapeutics to which we can point in the response to the COVID-19 pandemic, such as the introduction of mRNA vaccines and the accelerated path to its authorization through Operation Warp Speed. However, in anticipation of future known and unknown health security threats, including new pandemics, biothreats, and climate-related health emergencies, our answers need to be much faster, cheaper, and less disruptive to other operations. One path to a more permanent state of readiness is to create a commercial public health emergency payment system to use the full power of commercial healthcare reimbursement, providing clear and tunable market signals to catalyze investment in anticipation and in response to public health emergencies.
Challenge and Opportunity
There are two key problems we have yet to solve. First, how do we break out of the panic/neglect cycle of investments in emergency medical countermeasures (MCMs), i.e invest more in prevention and preparedness than in response? Secondly, how can we avoid any friction in capital (both public and private) once there is an emergency, while preserving the ability to fine tune incentives as needs evolve?
Many of the most promising and impactful tools applicable to emerging infectious diseases and public health emergency (PHE) management more broadly (e.g. wastewater surveillance, home testing, vaccinations, improved indoor air quality), face strong headwinds due to small, poorly defined, and/or unstable markets, significantly reducing private investment in them and relying on seemingly stochastic public investments by a fragmented set of federal, state, and local agencies.
Additionally, there is a prevailing assumption within the health security community that it operates largely outside the commercial U.S. healthcare system due to lack of private incentives (as opposed to, for instance, the development and use of cancer therapies). This, however, reflects a policy decision and not fundamental underlying market demand. And yet there remain two key realities to contend with: pandemic-related demand is largely unpredictable, and we have, thus far, been unable to effectively amortize pandemic costs into interpandemic periods.
U.S. health care insurers process more than nine billion claims for payment each year – a process which features a sophisticated, standardized accounting system that is widely understood by the entire healthcare industry; it is also a powerful signal of future market expectations that drives private and public R&D investment decisions.
Plan of Action
The U.S. government should create a prospective healthcare reimbursement code set that can anticipate the need for any product or service in the context of PHEs, including MCMs and infrastructure-based countermeasures. The goal would be to provide clear market signals and pull incentives, to encourage and accelerate development and appropriate utilization of medical countermeasures (diagnostics, vaccines, therapies, early warning detection, and others). This would complement other strategies, such as advanced R&D investments made by federal funding agencies including the Biomedical Advanced Research & Development Authority (BARDA). Creating clear reimbursement pathways would likely immediately attract private investment in MCMs in ways that are notoriously difficult currently and help enable rapid response to public health emergencies.
The development and management of this reimbursement system should be housed within the Centers for Medicare and Medicaid Services (CMS), and would require introduction of legislation clearly authorizing CMS to pay for products and services under EUA and use of prospective payment policies.
There are a variety of additional benefits to using a payment system like this for emergency response. This system could be a unique and core source of surveillance data, through conditions of payment policies, that can be used to provide intelligence and manage evolving emergencies such as outbreaks and pandemics, significantly reducing the need for additional data collection systems – need which proved to be a major bottleneck during COVID-19. Prospective codesets are not common in public and private insurance, but the existence of them would likely serve as a new and powerful tool for private investment in a capability that would be certain to have public health benefit were it to exist.
We propose that reimbursable services be categorized into three tiers.
Tier 1. Existing and prospective medical products/services under FDA EUA
This is the set of regulated medical products that would typically be considered for emergency use authorization by the FDA. It can include infectious disease diagnostics, vaccinations, and therapeutics. It can even include the use of repurposed generic drugs to mitigate potential drug shortages.
Tier 2. Existing public health products/services typically not requiring FDA approvals, but may have other regulatory hurdles
There are a variety of products and services not considered medical products but nonetheless play critical roles not only in response but in prevention of public health emergencies. Many of these technologies struggle to find stable markets in which to operate and are relegated to the sidelines. This includes genomic surveillance, remnant sample sequencing, or other surveillance testing capabilities, early warning systems, use of commercial wearables as a check engine light for possible infection prior to symptom onset. Many of these technologies need to be “always on” to be most effective.
Tier 3. Prospective public health products/services typically not requiring FDA approvals, but may have other regulatory hurdles
This category deviates from current models of healthcare reimbursement, because it is comprised of interventions that are carried out by service providers outside of healthcare delivery, but which nonetheless have high impact potential. This can include indoor air quality upgrades, or wastewater detection. Again, the commercialization of indoor air quality is in part impeded by poorly defined markets.
Value-based care models
While the above items are largely “fee for service” payment models, we can also envision the use of value-based care models here, focusing on community outcomes and providing flexibility to communities or other systems to achieve them. This can include models to prevent hospitalization due to PHE (e.g. COVID-19), prevent community transmission (could be directed at local public health agencies or other agents), herd immunity vaccination rates, etc.
Conclusion
One of the biggest frustrations amongst the life science and medical device communities in the COVID-19 response was that the government was not clear about target product profiles and advanced market commitments for the full range of products and services needed. These types of systems, if implemented early, would have sent clear and powerful signals to the market that would have quickly unlocked the necessary private sector investment needed to expedite product development needed for the response. Using the already widely adopted and understood commercial healthcare reimbursement system to incentivize and pay for prospective emergency product development and delivery is a novel, powerful, and turnkey approach to pandemic preparedness.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
Creating an HHS Loan Program Office to Fill Critical Gaps in Life Science and Health Financing
We propose the establishment of a Department of Health and Human Services Loan Programs Office (HHS LPO) to fill critical and systematic gaps in financing that prevent innovative life-saving medicines and other critical health technologies from reaching patients, improving health outcomes, and bolstering our public health. To be effective, the HHS LPO requires an authority to issue or guarantee loans, up to $5 billion in total. Federally financed debt can help fill critical funding gaps and complement ongoing federal grants, contracts, reimbursement, and regulatory policies and catalyze private-sector investment in innovation.
Challenge and Opportunity
Despite recent advances in the biological understanding of human diseases and a rapidly advancing technological toolbox, commercialization of innovative life-saving medicines and critical health technologies face enormous headwinds. This is due in part to the difficulty in accessing sustained financing across the entire development lifecycle. Further, macroeconomic trends such as non-zero interest rates have substantially reduced deployed capital from venture capital and private equity, especially with longer investment horizons.
The average new medicine requires 15 years and over $2 billion to go from the earliest stages of discovery to widespread clinical deployment. Over the last 20 years, the earliest and riskiest portions of the drug discovery process have shifted from the province of large pharmaceutical companies to a patchwork of researchers, entrepreneurs, venture capitalists, and supporting organizations. While this trend has enabled new entrants into the biotechnology landscape, it has also required startup companies to navigate labyrinthine processes of technical regulatory guidelines, obtaining long-term and risk-friendly financing, and predicting payor and provider willingness to ultimately adopt the product.
Additionally, there are major gaps in healthcare infrastructure such as lack of adequate drug manufacturing capacity, healthcare worker shortages, and declining rural hospitals. Limited investment is available for critical infrastructure to support telehealth, rural healthcare settings, biomanufacturing, and decentralized clinical trials, among others.
The challenges in health share some similarities to other highly regulated, capital-intensive industries, such as energy. The Department of Energy (DOE) Loan Program Office (LPO) was created in 2005 to offer loans and loan guarantees to support businesses in deploying innovative clean energy, advanced transportation, and tribal energy projects in the United States. LPO has closed more than $40 billion in deals to date. While agencies across HHS rely primarily on grants and contracts to deploy research and development (R&D) funding, capital-intensive projects are best deployed as loans, not only to appropriately balance risk between the government and lendees but also to provide better stewardship over taxpayer resources via mechanisms that create liquidity with lower budget impact. Moreover, private-sector financing rates are subject to market-based interest rates, which can have enormous impacts on available capital for R&D.
Plan of Action
There are many federal credit programs across multiple departments and agencies that provide a strong blueprint for the HHS LPO to follow. Examples include the aforementioned DOE Loan Programs Office, which provides capital to scale large-scale energy infrastructure projects using new technologies, and the Small Business Administration’s credit programs, which provide credit financing to small businesses via several loan and loan matching programs.
Proposed Actions
We propose the following three actions:
- Pass legislation authorizing the HHS LPO to make or guarantee loans supporting the development of critical medicines that serve unmet or underserved public health needs.
- Establish the LPO as a staffing division within HHS.
- Appropriate a budget to support the staffing and operations of the newly created HHS LPO.
Scope
Similar to how DOE LPO services the priorities of the DOE, the HHS LPO would develop strategy priorities based on market gaps and public health gaps. It would also develop a rigorous diligence process to prioritize, solicit, assess, and manage potential deals, in alignment with the Federal Credit Reform Act and the associated policies set forth by the Office of Management and Budget and followed by all federal credit programs. It would also seek companion equity investors and creditors from the private sector to create leverage and would provide portfolio support via demand-alignment and -generation mechanisms (e.g., advance manufacturing commitments and advanced market commitments from insurers).
We envision several possible areas of focus for the HHS LPO:
- Providing loans or loan guarantees to amplify investment funds that use venture capital or other private investment tools, such as early-stage drug development or biomanufacturing capacity. While these funds may already exist, they are typically underpowered.
- Providing large-scale financing in partnership with private investors to fund major healthcare infrastructure gaps, such as rural hospitals, decentralized clinical trial capacity, telehealth services, and advanced biomanufacturing capacity.
- Providing financing to test new innovative finance models, e.g. portfolio-based R&D bonds, designed to attract additional capital into under-funded R&D and lower financial risks.
Conclusion
To address the challenges in bringing innovative life-saving medicines and critical health technologies to market, we need an HHS Loan Programs Office that would not only create liquidity by providing or guaranteeing critical financing for capital-intensive projects but address critical gaps in the innovation pipeline, including treatments for rare diseases, underserved communities, biomanufacturing, and healthcare infrastructure. Finally, it would be uniquely positioned to pilot innovative financing mechanisms in partnership with the private sector to better align private capital towards public health goals.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
The DOE LPO, enabled via the Energy Policy Act of 2005, enables the Secretary of Energy to provide loan guarantees toward publicly or privately financed projects involving new and innovative energy technologies.
The DOE LPO provides a bridge to private financing and bankability for large-scale, high-impact clean energy and supply chain projects involving new and innovative technologies. It also expands manufacturing capacity and energy access within the United States. The DOE LPO has enabled companies involving energy and energy manufacturing technologies to achieve infrastructure-scale growth, including Tesla, an electric car manufacturer; Lithium Americas Corp., a company supplying lithium for batteries; and the Agua Caliente Solar Project, a solar power station sponsored by NRG Solar that was the largest in the world at its time of construction.
The HHS LPO would similarly augment, guarantee, or bridge to private financing for projects involving the development and deployment of new and innovative technologies in life sciences and healthcare. It would draw upon the structure and authority of the DOE LPO as its basis.
The HHS LPO could look to the DOE LPO for examples as to how to structure potential funds or use cases. The DOE LPO’s Title 17 Clean Energy Financing Program provides four eligible project categories: (1) projects deploying new or significantly improved technology; (2) projects manufacturing products representing new or significantly improved technologies; (3) projects receiving credit or financing from counterpart state-level institutions; and (4) projects involving existing infrastructure that also share benefits to customers or associated communities.
Drawing on these examples, the HHS LPO could support project categories such as (1) emerging health and life science technologies; (2) the commercialization and scaling access of novel technologies; and (3) expanding biomanufacturing capacity in the United States, particularly for novel platforms (e.g., cell and gene therapies).
The budget could be estimated via its authority to make or guarantee loans. Presently, the DOE LPO has over $400 billion in loan authority and is actively managing a portfolio of just over $30 billion. Given this benchmark and the size of the private market for early-stage healthcare venture capital valued at approximately $20 billion, we encourage the creation of an HHS LPO with $5 billion in loan-making authority. Using proportional volume to the $180 million sought by DOE LPO in FY2023, we estimate that an HHS LPO with $5 billion in loan-making authority would require a budget appropriation of $30 million.
The HHS LPO would be subject to oversight by the HHS Inspector General, OMB, as well as the respective legislative bodies, the House of Representatives Energy and Commerce Committee and the Senate Health, Education, Labor and Pension Committee.
Like the DOE LPO, the HHS LPO would publish an Annual Portfolio Status Report detailing its new investments, existing portfolio, and other key financial and operational metrics.
It is also possible for Congress to authorize existing funding agencies, such as BARDA, the Advanced Research Projects Agency for Health (ARPA-H), or the National Institutes for Health (NIH), with loan authority. However, due the highly specialized talent needed to effectively operate a complex loan financing operation, the program is significantly more likely to succeed if housed into a dedicated HHS LPO that would then work closely with the other health-focused funding agencies within HHS.
The other alternative is to expand the authority for other LPOs and financing agencies, such as the DOE LPO or the U.S. Development Finance Corporation, to focus on domestic health. However, that is likely to create conflicts of priority given their already large and diverse portfolios.
The project requires legislation similar to the Department of Energy’s Title 17 Clean Energy Financing Program, created via the Energy Policy Act of 2005 and subsequently expanded via the Infrastructure Investment and Jobs Act in 2021 and the Inflation Reduction Act in 2022.
This legislation would direct the HHS to establish an office, presumably a Loan Programs Office, to make loan guarantees to support new and innovative technologies in life sciences and healthcare. While the LPO could reside within an existing HHS division, the LPO would most ideally be established in a manner that enables it to serve projects across the full Department, including those from the National Institutes of Health, Food and Drug Administration, Biomedical Advanced Research and Development Authority, and the Centers for Medicare and Medicaid Services. As such, it would preferably not reside within any single one of these organizations. Like the DOE LPO, the HHS LPO would be led by a director, who would be directed to hire the necessary finance, technical, and operational experts for the function of the office.
Drawing on the Energy Policy Act of 2005 that created the DOE LPO, enabling legislation for the HHS Loan Programs office would direct the Secretary of HHS to make loan guarantees in consultation with the Secretary of Treasury toward projects involving new and innovative technologies in healthcare and life sciences. The enabling legislation would include several provisions:
- Necessary included terms and conditions for loan guarantees created via the HHS LPO, including loan length, interest rates, and default provisions;
- Allowance of fees to be captured via the HHS LPO to provide funding support for the program; and
- A list of eligible project types for loan guarantees.
Supporters are likely to include companies developing and deploying life sciences and healthcare technologies, including early-stage biotechnology research companies, biomanufacturing companies, and healthcare technology companies. Similarly, patient advocates would be similarly supportive because of the LPO’s potential to bring new technologies to market and reduce the overall Weighted Average Cost of Capital (WACC) for biotechnology companies, potentially supporting expanded access.
Existing financiers of research in biomedical sciences technology may be neutral or ambivalent toward this policy. On one hand, it would provide expanded access to syndicated financing or loan guarantees that would compound the impact of each dollar invested. On the other hand, most financiers currently use equity financing, which enables the demand for a high rate of return via subsequent investment and operation. An LPO could provide financing that requires a lower rate of return, thereby diluting the impact of current financiers in the market.
Skeptics are likely to include groups opposing expansions of government spending, particularly involving higher-risk mechanisms like loan guarantees. The DOE LPO has drawn the attention of several such skeptics, oftentimes leading to increased levels of oversight from legislative stakeholders. The HHS LPO could expect similar opposition. Other skeptics may include companies with existing medicines and healthcare technologies, who may be worried about competitors introducing products with prices and access provisions that have been enabled via financing with lower WACC.
Slow Aging, Extend Healthy Life: New incentives to lower the late-life disease burden through the discovery, validation, and approval of biomarkers and surrogate endpoints
The world is aging. Today, some two thirds of the global population is dying from an age-related condition. Biological aging imposes significant socio-economic costs, increasing health expenses, reducing productivity, and straining social systems. Between 2010 and 2030, Medicare spending is projected to nearly double – to $1.2 trillion per year. Yet the costly diseases of aging can be therapeutically targeted before they become late-stage conditions like Alzheimer’s. Slowing aging could alleviate these burdens, reducing unpaid caregivers, medical costs, and mortality rates, while enhancing productivity. But a number of market failures and misaligned incentives stand in the way of extending the healthy lifespan of aging populations worldwide. New solutions are needed to target diseases before they are life-threatening or debilitating, moving from retroactive sick-care towards preventative healthcare.
The new administration should establish a comprehensive framework to incentivize the discovery, validation, and regulatory approval of biomarkers as surrogate endpoints to accelerate clinical trials and increase the availability of health-extending drugs. Reliable biomarkers or surrogate endpoints could meaningfully reduce clinical trial durations, and enable new classes of therapeutics for non-disease conditions (e.g., biological aging). An example is how LDL (a surrogate marker of heart health) helped enable the development of lipid-lowering drugs. The current lack of validated surrogate endpoints for major late-life conditions is a critical bottleneck in clinical research. Because companies do not capture the majority of the benefit from the (expensive) validation of biomarkers, the private sector under-invests in biomarker and surrogate endpoint validation. This leads to countless lives lost and to trillions of public dollars spent on age-related conditions that could be prevented by better-aligned incentives. It should be an R&D priority for the new administration to fund the collection and validation of biomarkers and surrogate endpoints, then gain regulatory approval for them. As we explain below, the existing FNIH Biomarkers Consortium does not fill this role.
Currently, companies are understandably hesitant to invest in validation without clear rewards or regulatory pathways. The proposed framework would encourage private companies and laboratories to contribute their biomarker data to a shared repository. This repository would expedite regulatory approval, moving away from the current product-by-product assessment that discourages data sharing and collaboration. Establishing a broader pathway within the FDA for standardized biomarker approval would allow validated biomarkers to be recognized for use across multiple products, reducing the existing incentives to safeguard data while increasing the supply of validated biomarkers and surrogate endpoints. Importantly, this would accelerate the development of drugs which holistically extend the healthspan of aging populations in the U.S. by preventing instead of treating late-stage conditions. (Statins similarly helped prevent millions of heart attacks.)
Key players such as the FDA, NIH, ARPA-H, and BARDA should collaborate to establish a streamlined pathway for the collection and validation of biomarkers and surrogate endpoints, allowing these to be recognized for use across multiple products. This initiative aligns with the administration’s priorities of accelerating medical innovation and improving public health with the potential to add trillions of dollars in economic value by making treatments and preventatives available sooner. This memo outlines a framework applicable to various diseases and conditions, using biological aging as a case study where the validation of predictive and responsive biomarkers may be vital for significant breakthroughs. Other critical areas include Alzheimer’s disease and amyotrophic lateral sclerosis (ALS), where the lack of validated surrogate endpoints significantly hinders the development of life-saving and life-improving therapies. By addressing these bottlenecks, we can unlock new avenues for medical advancements that will profoundly improve public health and mitigate the fast-growing, nearly trillion-dollar Medicare spend on late-life conditions.
Challenge and Opportunity
By 2029, the United States will spend roughly $3 trillion dollars yearly – half its federal budget – on adults aged 65 and older. A good portion of these funds will go towards Medicare-related expenses that could be prevented. Yet the process of bringing preventative drugs to market is lengthy, costly, and currently lacking in commercial incentives. Even for therapeutics that target late-stage diseases, drug development often takes 10+ years and cost estimates range between $300 million to $2.8 billion. This extensive duration and expense are due, in part, to the reliance on traditional clinical endpoints, which require long-term observation and longitudinal data collection. The burden of chronic diseases is growing, and better biomarkers and surrogate endpoints are needed to accelerate the development of therapeutics that prevent non-communicable diseases and age-related decline. Chronological age, for instance, is a commonly used but inadequate surrogate marker for biological age. This means that, to date, clinical trials on therapeutics designed to improve the biology of aging take decades to validate, rather than years. As a result, pharmaceutical companies find more short-term rewards in treating late-stage diseases, since developing drugs that reduce overall age-related decline requires longer and currently uncertain endpoints.
The validation of reliable biomarkers and surrogate endpoints offers a promising solution to this challenge. Biological measures often correlate with and predict clinical outcomes, and can therefore provide early indications of whether a treatment is effective. If sufficiently predictive, biomarkers can serve as surrogate clinical endpoints, potentially reducing the duration and cost of clinical trials. Validated biomarkers must accurately predict clinical outcomes and be accepted by regulatory authorities, yet the validation process is underfunded due to insufficient commercial incentives for individual agents to share their biomarkers to be used as a public good. (From a purely financial standpoint, companies are better off targeting diseases with known endpoints.)
The most prominent existing efforts to advance biomarkers and surrogate endpoints are the Foundation for the National Institute of Health’s (FNIH) Biomarkers Consortium and the FDA’s Biomarker Qualification Program. Established in 2006, the Biomarkers Consortium is a public-private partnership aimed at advancing the development and use of biomarkers in medical research. Meanwhile, the FDA’s qualification program was the result of the 21st Century Cures Act, passed in 2016, which underscored the critical role biomarkers play in accelerating medical product development. The Act mandated the FDA to implement a more transparent and efficient process for biomarker qualification.
Despite the Consortium’s ambitious goals, the rate of biomarker qualification by the FDA has been slow. Since its inception in 2006, only a small number of biomarkers have been successfully qualified. This sluggish progress has been a source of criticism for stakeholders, especially given the high level of resources and collaboration involved. For example, the process of validating biomarkers for osteoarthritis under the Consortium’s “PROGRESS OA” project has been ongoing since Phase 1 and still faces hurdles before full qualification. We are of the view that this is the result of two issues. Firstly, the qualification process, which involves FDA approval, is seen as overly complex and time-consuming. Despite the 21st Century Cures Act aiming to streamline the process, resulting in the qualification pathway, it remains a significant challenge. The difficulty in navigating the regulatory landscape can limit the impact of Biomarkers Consortium (BC) projects. The Kidney Safety Project, for example, faced substantial regulatory hurdles before finally achieving the first qualification of a clinical safety biomarker. Secondly, even though the Consortium operates in a precompetitive space, there are ongoing challenges related to data sharing. Companies may still hesitate to share critical data that could advance biomarker validation out of concern for losing a competitive edge, which hampers collaboration. To address these issues, it is crucial to implement a framework that promotes data sharing in the academic and private sectors, providing strong incentives for the validation and regulatory approval of biomarkers, while improving regulatory certainty with a standardized regulatory process for surrogate endpoint validation.
The current boom in biotechnology underscores the urgency of addressing persisting inefficiencies. Without changes, we face a significant bottleneck in proving the efficacy of new drugs. This is exacerbated by Eroom’s Law—the observation that drug discovery is becoming slower and more expensive over time. This growing inefficiency threatens to hinder the development of new, life-saving treatments at a time when the American population is aging and rapid medical advancements are crucial to deter increasing medical and social costs. In just 11 years—between 2018 and 2029—the U.S. mandatory spending on Social Security and Medicare will more than double, from $1.3 trillion to $2.7 trillion per year. Yet the costly diseases of aging can be therapeutically targeted before they become late-stage conditions like Alzheimer’s. For federal policymakers, taking immediate action to improve data sharing and biomarker validation processes is vital. Failure to do so will not only stifle innovation but also delay the availability of critical therapies that could save countless lives and accelerate economic growth in the long run. Prompt policy intervention is essential to capitalize on the current advancements in biotechnology and ensure the development of new life-saving tests, tools, and drugs.
Implementing pull-incentives for data sharing now can help the United States adjust to its new demographic structure, where adults in advanced age prevail, while fertility rates decline. It can also mitigate the escalating costs and timelines of clinical trials, and accelerate the approval of life-saving, health-extending drugs. If our proposed framework is successfully implemented, a robust pool of biomarker data will be established, significantly facilitating the discovery and validation of biomarkers. This will result in several key advancements, including shortened clinical trial durations, increased R&D investment, faster drug approvals, and even increased drug efficacy. Additionally, new drug classes targeting non-disease endpoints, such as biological aging, could be developed. Just as the discovery of LDL as a surrogate marker of heart health was critical in enabling the testing and development of statins, the discovery of clinical-grade biomarkers may unlock new therapeutics designed to target the mechanisms that drive human aging, slowing down the progression of age-related diseases (like cancers) before they become deadly and socio-economically expensive.
Plan of Action
To address the challenge of inefficient data sharing, validation, and approval of biomarkers, we propose implementing a series of pull-incentives aimed at encouraging pharmaceutical companies to contribute their relevant biomarker data to a shared repository and undertake the necessary research and analysis for public validation. These validated biomarkers can then be formally accepted by regulators as surrogate endpoints for drug approval, accelerating the drug development process and reducing late-life costs.
Recommendation 1. An NIH-FDA initiative for Biomarkers and Surrogate Endpoints Within the NIA
Most existing agencies focus on single, often late-stage diseases. This is at odds with a holistic understanding of human biology. A new initiative within the National Institute on Aging (NIA) could be devoted to the discovery, collection, and validation of biomarkers and surrogate endpoints for overall human health and age-related decline. Most National Institutes of Health funds are currently devoted to the diseases of aging (think cancers, Alzheimer’s, heart disease, or Parkinson’s.) Within the NIA, research on Alzheimer’s disease alone receives roughly eight times more funding than the biology of aging, with few human-relevant results. Every federal agency and U.S. individual would benefit from better biomarkers of long-term health and from an understanding of how to measure the biology of aging. Yet no single agent has the incentives to collect and validate this data, for instance by shouldering the costs of validating predictive and responsive biomarkers of aging.
This new initiative could also be devoted to the development of preclinical, human-relevant methodologies that could broadly facilitate or streamline drug development. In 2022, the FDA Modernization Act 2.0 approved the use of in vitro and in silico New Approach Methodologies (NAMs) like cell-based assays (e.g. organs-on-chips) or computer models (like virtual cells) in preclinical development to reduce or replace animal studies, especially “where no pharmacologically relevant animal species exists.” This may be the case for human aging, where no single animal model reflects the full complex biology of our aging process.
At present, these technologies cannot accurately represent the multifactorial processes of aging, and they cannot model entire organisms. Much work remains to be done to even understand how to “code” aging into organs-on-chips. Yet if supplemented by approaches like in vivo pooled screening, next generations of human-relevant in vitro or in silico methodologies (like virtual cells) could be infused with the complex data needed to accelerate clinical trial results and increase drug efficacy. For in vitro and in silico models to reproduce key aspects of aging biology, a better understanding of how human aging works in living organisms — and what markers to include to represent it either virtually or in vitro — may be needed. Yet pharmaceutical companies, startups, health insurance firms, and even research hospitals again lack the incentives to shoulder the costs of collecting and validating this type of data. This means a new office within a federal agency may be needed to supply these incentives.
Recommendation 2. New Data-sharing Incentives
The specific incentives used would need to be developed in collaboration with policymakers and industry stakeholders, but a few are outlined below:
Pull-incentives
One possibility is offering transferable Priority Review Vouchers (PRVs) or similar pull incentives to companies that share their biomarker data. PRVs are currently awarded by the FDA to companies developing drugs for neglected tropical diseases, rare pediatric diseases, or medical countermeasures. A PRV allows the holder to expedite the FDA review of another drug from 10 months to 6 months, and holds significant financial value. Offering transferable PRVs for drugs designed to target biological aging, for instance, could create the incentives needed for pharmaceutical companies to target early-stage age-related conditions before they turn into diseases.
The creation of a new PRV category would require legislative action. Our proposed NIH-FDA initiative would be well positioned to oversee the issuance of PRVs, working with government agencies and think tanks to determine, for instance, what an “aging therapeutic” means, and what a company needs to achieve to gain a PRV for a longevity drug. The Alliance for Longevity Initiatives, for instance, has developed an advanced approval pathway for health-extending drugs that directly target the biology of aging. Another possible strategy would be for the FDA to encourage drugs that target multiple disease indications at once, perhaps offering discounts or incentives for every extra biomarker or surrogate endpoint validated. This could effectively encourage the development of drugs that do more than marginally improve on existing interventions.
We acknowledge that an overabundance of PRVs can saturate the market, decreasing their value and weakening the intended pull-incentive for pharmaceutical innovation. A response would be to demand that proposals to issue additional PRVs include a comprehensive market impact analysis to mitigate unintended economic consequences. Expanding the number of PRVs can also place extra demands on the FDA’s limited resources, potentially leading to longer approval times for other essential medications, even though PRV holders often delay redemption, preventing an immediate influx of priority review applications. The PRV system may inadvertently favor larger, well-established pharmaceutical companies that have the means to acquire and leverage PRVs effectively, creating barriers for smaller firms and startups. These are all spill-over problems worth solving for the potential upshot of mitigating late-life disease costs and encouraging drugs that holistically improve the human healthspan.
Biomarker Data Sharing as a Condition of Federal Funding
Federal funding recipients are legally obligated to make their research publicly accessible through agency-specific policies aimed at advancing open science. This mandate was strengthened by the 2022 OSTP Memorandum. Despite this clear mandate, the implementation of public access policies has been uneven across federal agencies, with progress varying due to differences in resources, technical infrastructure, and agency-specific priorities. The 2022 OSTP Public Access Memorandum aims to accelerate agency efforts to enhance public access infrastructure and policies. This updated guidance presents an opportunity for agencies to not only meet immediate data-sharing requirements but also to expand policy scopes to include essential clinical data, such as biomarker data from clinical trials. To meet these goals, agencies should ensure that funding agreements explicitly require the publication of comprehensive biomarker data and that suitable repositories are available to store and share these critical datasets effectively.
Case Study: Project NextGen
A prime example of the potential success of such initiatives is Project NextGen, a program led by BARDA in collaboration with the NIH to advance the next generation of COVID-19 vaccines and treatments. As part of its vaccine program, Project NextGen includes centralized immunogenicity assays with the overarching goal of establishing correlates of protection, which could serve as surrogate biomarkers for next-gen vaccines. These assays are collected during Phase 2b vaccine studies sponsored by Project NextGen, which have been designed to measure a number of secondary immunogenicity endpoints including systemic and mucosal immune responses. Developers share their assays so that they can be used as a public good, in return for federal funding. This effort demonstrates the feasibility and benefits of a federally led effort to share assay data to advance biomarker validation and drug development.
Recommendation 3. Create and Manage a Data Repository
To enhance collaborative research and ensure the efficient use of publicly funded clinical data, we recommend establishing a secure data repository. This repository will serve as a centralized platform for data submission, storage, and access. Management of the repository could be undertaken by a federal agency, such as the NIH, leveraging their experience with the Biomarkers Consortium, perhaps in partnership with non-governmental organizations like the Biomarkers of Aging Consortium. Drawing from existing models, such as Project NextGen’s assay data management, can provide valuable insights into the implementation and operationalization of the repository.
The cost of establishing and maintaining this repository, including data storage, management, and access controls, would be dwarfed by the socio-economic returns it could provide. This repository can facilitate data sharing, protect sensitive information, and promote a collaborative environment that accelerates biomarker validation and approval, while ensuring pharmaceutical companies that their hard-earned data is safely stored.
The securely stored data in the repository would primarily be accessible to qualified researchers, clinicians, and policymakers involved in biomarker research and development, including academic researchers, pharmaceutical companies, and public health agencies. Access would be granted through an application and review process. The benefits of this repository are multifaceted: it accelerates research by providing a centralized database, enhances collaboration among scientists and institutions, increases efficiency by reducing redundancy and improving data management, ensures data security through robust access controls, offers cost-effectiveness with long-term socio-economic returns, and supports regulatory bodies with comprehensive data sets for more informed decision-making.
Recommendation 4. Create A Regulatory Pathway with Broader Application
To accelerate the adoption of validated biomarkers and surrogate endpoints in drug development, we propose the creation of a streamlined regulatory approval process within the FDA. This new pathway would establish clear criteria and standardized procedures for biomarker evaluation and approval, facilitating their recognition for use across multiple products and therapeutic areas.
Currently, the FDA’s Center for Drug Evaluation and Research (CDER) operates the Biomarker Qualification Program (BQP), which allows drug developers to seek regulatory qualification for specific contexts of use. While this program fosters collaboration between the FDA and external stakeholders, biomarkers are qualified on a case-by-case basis, limiting their broader applicability across different drug development programs.
Additionally, the FDA maintains a Table of Surrogate Endpoints that have been used as the basis for drug approvals under the accelerated approval pathway. However, this table primarily serves as a reference and does not comprehensively address the need for a streamlined approval process for biomarkers and surrogate endpoints.
By developing a framework that moves away from traditional product-by-product assessments, the FDA could reduce existing barriers to biomarker and surrogate endpoint discovery and approval. This approach would encourage data sharing and collaboration among pharmaceutical companies and research institutions, leading to faster validation and broader acceptance of these critical tools in drug development.
This proposal builds upon existing legislative efforts, such as the 21st Century Cures Act of 2016, which includes provisions to accelerate medical product development and supports the use of biomarkers and surrogate endpoints in the regulatory process. Furthermore, it aligns with the FDA’s ongoing efforts to provide clarity on evidentiary criteria for biomarker qualification, as outlined in the 2018 guidance document “Biomarker Qualification: Evidentiary Framework.”
Inspiration for this approach can be drawn from the Advanced Approval Pathway for Longevity Medicines (AAPLM) proposed by the Alliance for Longevity Initiatives (See AAPLM-Whitepaper). The AAPLM includes provisions such as a special approval track, a priority review voucher system, and indication-by-indication patent term extensions, which align economic incentives with the transformative health improvements that longevity medicines can provide. These measures offer a valuable template for facilitating the recognition and approval of biomarkers. Adding to the existing FDA table of surrogate endpoints that can serve as the basis for drug approval or licensure, and referencing existing collaborations between the NIH and FDA, such as the Biomarkers Consortium, can provide a robust foundation for new biomarker evaluations. Ultimately, this regulatory innovation will support the development of life-saving drugs, enhance public health outcomes, and meaningfully contribute to economic growth by bringing effective treatments to market more quickly.
Conclusion
Today, over two thirds of all deaths in the United States are the result of an age-related condition. The burden of non-communicable diseases is growing, and better biomarkers and surrogate endpoints are needed to target diseases before they are life-threatening or debilitating. The next administration should implement a comprehensive framework to promote data sharing and incentivize the validation and regulatory approval of biomarkers and surrogate endpoints. This aligns directly with the administration’s goal to make Americans healthy. These solutions can substantially reduce the duration and cost of clinical trials, accelerate the development of life-saving drugs, and improve public health outcomes. It is possible and necessary to create an environment that encourages and rewards pharmaceutical companies to share crucial data that accelerates medical innovation. By discovering and validating predictive and responsive biomarkers of health and disease, new therapeutic classes can be developed to directly target biological aging and prevent most forms of cancers, heart disease, frailty, vulnerability to severe infection, and Alzheimer’s. This will enable the United States to remain at the forefront of medical research, and to respond to the growing demographic crisis of aging populations in declining health.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
A number of market failures stand in the way of the discovery and validation of predictive, reliable, and responsive biomarkers. First, it’s currently expensive to test drugs in multiple disease indications, which means pharmaceutical companies are often incentivized to focus on late-stage diseases (e.g. delaying death by a terminal cancer by three months), since this drug class is more easily and quickly trialed. The FDA also strongly assumes that a treatment ought to modulate a single outcome. (Think life/death; heart disease/no heart disease.) Therapeutics that target biological aging, for instance, would take decades to test without validated biomarkers or widely accepted surrogate endpoints.
Aging research, for instance, has seen a 70-fold increase in venture capital funding since the last decade. Yet so far—and this is a critical asterisk—misaligned commercial incentives have mostly optimized for unproven supplements, imprecise biological-age-tracking apps, and unsafe experimental therapies or cosmetics. The most well-meaning investors and founders in “longevity” often end up developing drugs for single disease indications (like osteoarthritis, or obesity) to avoid bankruptcy or as a path to self-fund their intent of developing drugs that more holistically target the mechanisms that drive aging. arket incentives need to be aligned to the pressing social needs these therapeutics could respond to.
The federal government is uniquely positioned to coordinate large-scale initiatives that require significant resources and regulatory oversight. While private sector companies play crucial roles in drug development, they often lack the incentives to self-coordinate and the authority to drive comprehensive data-sharing and biomarker validation efforts.
Cohesion from data-collection to regulatory approval of biomarkers is going to be key if surrogate endpoints are actually going to be adopted. Having the federal government oversee all stages will ensure this cohesion.
The Biomarkers Consortium has made meaningful strides in advancing biomarker research, but they have not succeeded in acquiring sufficient data. The consortium relies on voluntary, precompetitive collaboration without providing strong financial or legislative incentives for data sharing. It does not maintain a centralized, secure data repository, and struggles with fragmented data sharing. It also lacks influence over the FDA’s biomarker qualification process, which remains complex and time-consuming. This has resulted in slow progress due to hesitancy from private entities to share valuable data. Our solution differs by directly addressing this data-sharing hurdle through a series of targeted incentives that reduce the case-by-case assessment currently required, and enable broader application of validated biomarkers across multiple drugs and therapeutic areas.
By introducing legislative changes to authorize patent extensions and expand Priority Review Vouchers (PRVs), we create compelling reasons for companies to share their data. Additionally, our proposal includes the development of a centralized data repository with a streamlined regulatory approval process, inspired by the Advanced Approval Pathway for Longevity Medicines (AAPLM). This approach not only incentivizes data sharing but also provides a clear and efficient pathway for biomarker validation and regulatory acceptance. By leveraging existing frameworks and offering tangible rewards, our solution proposes an increase in incentives, to match the socioeconomic benefits that may be unlocked by more accessibility to the wealth of existing but undersupplied biomedical data.
The FDA’s Accelerated Approval Pathway is indeed a valuable tool that allows for the approval of drugs based on surrogate endpoints that are reasonably likely to predict clinical benefit. This pathway requires substantial evidence showing that these surrogate endpoints are linked to clinical outcomes, usually gathered from rigorous clinical trials. However, it typically applies to surrogate endpoints validated for specific uses or products. Our goal is to establish a new pathway that supports the validation and use of surrogate endpoints across multiple products. By validating biomarkers that can be used across various drugs, we can streamline the drug development process, reducing the time and cost associated with bringing new therapies to market. This broader approach would enhance efficiency, reduce drug development time and costs, and promote innovation by encouraging pharmaceutical companies to invest in research, knowing that successful biomarkers can have wide-reaching applications.
Pharmaceutical companies could push back on this proposal due to concerns over losing their competitive advantage by sharing proprietary data. They might reasonably fear that sharing valuable biomarker data could erode their market position and intellectual property. By involving pharmaceutical companies in the development of the proposal, we can better understand their concerns and tailor incentives accordingly. One effective strategy would be to offer significant financial incentives, such as Priority Review Vouchers (PRVs) or patent term extensions to companies that share their data. These incentives can offset the perceived risks and provide tangible benefits that make data sharing more attractive. By making PRVs transferable and offering additional incentives to small biotechnology companies, this policy can be implemented without overly favoring large pharmaceutical companies. Another possible strategy would be for the FDA to encourage drugs that target multiple disease indications at once, perhaps offering discounts or incentives for every extra biomarker or surrogate endpoint validated. Fostering a collaborative environment where the benefits of shared data (such as accelerated drug approvals and reduced R&D costs) are clearly communicated can reduce hurdles. Engaging economists to quantify the long-term economic gains to individual pharmaceutical companies as well as to society, while demonstrating how shared data can lead to industry-wide advancements, can further encourage participation. By providing competitive enough incentives, a framework can be created that balances the interests of pharmaceutical companies with the broader goal of advancing medical innovation and public health.
The first step to get this proposal off the ground is to introduce legislative changes that authorize patent extensions and expand the eligibility for Priority Review Vouchers (PRVs). These legislative changes will create the necessary incentives for pharmaceutical companies to participate in the program by offering tangible benefits that offset the risks associated with data sharing.
Simultaneously, developing and launching a pilot program for the centralized data repository is crucial. This pilot should focus on a specific subset of biomarkers for high-priority diseases and non-disease indications to demonstrate the feasibility and benefits of the proposed framework. By starting with a targeted approach, we can gather initial data, test the processes, and make any necessary adjustments before scaling up the program. This pilot will not only help in garnering support from stakeholders by showcasing the practical benefits of the framework but also refine the approach based on real-world feedback, ensuring a smoother and more effective broader implementation.
Similar efforts in the past have often been hindered by a lack of incentives for data sharing and collaboration, along with fragmented regulatory processes. Our proposal aims to overcome these obstacles by introducing strong incentives which will encourage companies to share their data. Moreover, we propose creating a standardized regulatory pathway for biomarker approval, which will streamline the process and reduce fragmentation. By involving key federal agencies, we ensure a coordinated and comprehensive implementation, thus avoiding the pitfalls that have doomed past efforts.
The status quo is unacceptable. Millions of lives are lost or debilitated every year due to the slow and costly process of bringing new drugs to market, which is hindered by the lack of validated biomarkers and surrogate endpoints. The recommended course of action leverages existing regulatory frameworks and incentives that have proven effective in other contexts, such as the use of Priority Review Vouchers (PRVs) for neglected tropical diseases. By adapting these mechanisms to encourage data sharing and biomarker validation, we can build on established successes while addressing the specific challenges of the current drug development landscape.
This approach ensures that we utilize proven strategies to accelerate drug development and approval, reducing the overall time and cost associated with clinical trials. By fostering a collaborative environment and providing tangible incentives, we can significantly enhance the efficiency and effectiveness of the drug development process. This targeted strategy not only addresses the immediate needs but also sets a foundation for continuous improvement and innovation in the field of medical research, ultimately saving lives and improving public health outcomes.
Establishing a National Water Technology Pipeline
The next administration should establish a National Water Technology (Pipeline) to spur the innovation and commercialization of water technologies. The Pipeline should be designed to:
- Proactively deploy broad-spectrum monitoring and treatment technologies nationwide to avoid the devastating societal impacts of water contaminants.
- End significant sanitary sewer overflows that pose risks to human and environmental health.
- Ensure that every community in America has access to affordable and safe drinking water.
A National Water Technology Pipeline would mobilize American entrepreneurs and manufacturers to lead on research and development of the next generation of solutions in water treatment, monitoring, and data management. The Pipeline would facilitate commercialization of later-stage water technologies by identifying innovative next-to-market solutions, proving technology through competitive demonstration projects, and deploying market-ready technology at full scale with federal funding support. An underlying objective of the Pipeline would be to improve water quality and access in the United States while addressing mounting infrastructure and maintenance costs. The Pipeline would also place an emphasis on training the next generation of technology-focused water professionals and strengthening community engagement and customer service.
Modernizing the water sector will require the federal government to renew its commitment to investing in water. In recent years, the water sector received only 4% of its funding from the federal government: a far lower fraction than other infrastructure sectors, such as highways (25%), mass transit and rail (23%), and aviation (45%). The funding injection from the Bipartisan Infrastructure Law (BIL) has provided a temporary step-change in federal investment, but the substantial gap in funding is still anticipated to grow. Increasing federal funding for water technology advancement even by a percentage point would have hugely beneficial impacts. By dedicating 1% of projected water infrastructure costs for a “good state of repair”—an estimated $12 billion over the next 10 years—the next administration can build a robust National Water Technology Pipeline, ushering in a new era of water and sanitation technologies. A similar scale of investment, $25 billion over 10 years for clean energy demonstrations, was authorized through BIL for the Department of Energy (DOE).
Challenge and Opportunity
The next administration will inherit water and wastewater infrastructure that the American Society of Civil Engineers has given a C- and D+ rating, respectively in 2021, which is essentially unchanged from the prior D and D+ rating in 2017. Much of the water and wastewater infrastructure across the United States is more than half a century old. These infrastructure assets are showing signs of significant deterioration and displaying strong risks of failure as they approach the end of their service lives. Put simply, many U.S. water systems are not equipped to handle emerging treatment requirements and increasing severe weather challenges.
We cannot address our nation’s water infrastructure crisis without addressing water infrastructure funding for modern systems. One problem is that federal water infrastructure funding has simply dried up. In the 1970s and early 1980s, federal funding accounted for 15–30% of water infrastructure funding nationwide. This fraction has recently declined to a baseline of only 4%, far lower than other infrastructure sectors. Municipalities have been forced to raise local water rates to cover the funding gap. Access to adequate supplies of clean water is quickly becoming unaffordable for many Americans as a result, and recent polls show that the percentage of voters who find their water service unaffordable is on the rise. The inadequacy of current investment in water is both perception and reality.
A second problem is the growing cost of operating and maintaining water infrastructure. Nearly three-quarters of the public spending in the water sector supports operations and maintenance water systems, often legacy facilities. EPA conservatively estimates expenditures of $1.2 trillion over the next 20 years are needed just to maintain legacy drinking water ($625B) and wastewater ($630B) systems at current levels of service, without any modernization. Moreover, the U.S. water sector is large and complex, including over 50,000 community water systems and 16,000 sanitary sewer systems nationwide. Such a balkanized system makes it difficult to transfer innovative experiences and funding strategies across jurisdictional boundaries.
These challenges were exacerbated by the financial stresses and widespread supply chain challenges that the COVID-19 pandemic placed on municipalities. COVID-19 highlighted water treatment as an essential service. Serving as one of the most important tools we have for reducing the spread of infectious disease, clean water merits robust federal investment. COVID-19 also highlighted the usefulness of wastewater collection systems as early warning detection systems via wastewater-based epidemiology. Had there been investment in these wastewater based monitoring systems over the previous decade, the tracking of COVID-19 would have been significantly improved. The water industry is also vital to operations of other sectors essential to human health, environmental health, energy production, and transportation. Every dollar invested in drinking water and wastewater infrastructure increases GDP by $6.35, creates 1.6 new jobs, and provides $23 in public health-related benefits. Investing in the water sector is investing in the U.S. economy. The United States is lagging behind many other countries in dealing with issues as wide ranging as water-loss reduction, asset management, customer engagement, and customer service. It is past time to catch up.
The next administration should view these challenges as opportunities. Much has changed for the water sector in the last four years including the rise to prominence of artificial intelligence and machine learning capabilities, increased availability of low-cost sensors, widespread use of wastewater-based epidemiology as an early warning system for health risks, and rising challenge of malevolent cybersecurity threats from bad actors. Instead of propping up aging facilities, the next administration can incentivize investment into—and demonstration of—the most advanced and efficient water systems. Moreover, the next administration can incentivize the deployment of advanced monitoring and analytic technologies to proactively evaluate equipment health and develop a data-driven schedule for infrastructure replacement instead of ad hoc upgrades. The recent BIL funding provides an example of a historic $15B water infrastructure investment to replace lead pipes nationwide. While some innovative construction methods are being utilized, the water sector still lacks a suitable technology solution for the non-invasive identification of buried lead pipes, this is exactly the type of challenge a Technology Pipeline could address. Establishing a National Water Technology Pipeline will help unify our nation’s water sector and create pathways to expedite the installation of modern systems instead of maintaining outdated legacy technologies.
The next administration should empower the National Institute of Standards and Technology (NIST) in the Department of Commerce to lead this Pipeline development. (Isn’t the Environmental Protection Agency (EPA) in charge of water quality for the United States? See FAQs.) With the recent expansion of capabilities afforded by the CHIPS and Science Act, NIST is better positioned now more than ever to facilitate validation of new breakthroughs in low-cost sensors, data management and analytics, internet of things (IOT), predictive analysis, and machine learning and other technologies that are proving capable of significant productivity gains for water utilities and can prevent catastrophic system failures. As an example, modern sensor and data solutions have been demonstrated to reduce the historical challenge of sanitary sewer overflows by optimizing existing sewer networks. Further advances could even enable real-time monitoring of lead, per- and polyfluoroalkyl substances (PFAS), and other contaminants. Operational and maintenance savings realized from these technological solutions can be directly reinvested into infrastructure needs and/or used to subsidize water costs for low-income customers. The growth of the semiconductor manufacturing sector and water-intensive microchip facilities are also closely linked to water and wastewater infrastructure and NIST can play a part in driving those solutions. NIST can also engage in interagency initiatives with the DOE to address specific water-energy nexus challenges
Plan of Action
The next administration should set the United States on a path to build the most advanced water systems in the world by launching a new National Water Technology Pipeline initiative. The Pipeline would accelerate adoption of existing “market-ready” solutions from around the world while also fostering the development of next-generation technologies by American innovators. The Pipeline should be structured around three main goals:
- Proactively deploying broad-spectrum monitoring and treatment technologies nationwide to avoid the devastating societal impacts of water contaminants.
- Ending significant sanitary sewer overflows that pose risks to human and environmental health.
- Ensuring that every community in America has access to affordable and safe drinking water.
Achieving these goals will require a multi-pronged approach, as described below.
New Public-Private Frameworks to Enable Innovation
NIST should lead a new collaborative effort aligned with the Department of Commerce’s mission to “promote U.S. innovation and industrial competitiveness” in the water sector. Specifically, NIST should partner with industry leaders from utilities, vendors, equipment manufacturers, and system designers to develop a unified framework for deploying new technologies in the water sector. The framework would include verification and provisional standards to allow utilities to more easily adopt new technologies and to open the market for private investment in public water infrastructure. These efforts could be modeled after NIST efforts on the Manufacturing USA program or DOE’s Office of Clean Energy Demonstrations (OCED).
Technology Demonstration and Deployment Network
Congress should appropriate 1% of the projected total water infrastructure costs each year for the next 10 years to support water innovation. Specifically, these monies would be used for competitive grants (administered by NIST) to assist “early adopter” water utilities in deploying improved technologies, to fund state-based water innovation councils, and to support small manufacturers innovating in the water sector. NIST would coordinate with state, county, and local utilities, industry associations and professionals, and manufacturers to facilitate identification, testing, validation, and adoption of viable technology solutions. A similar development and dissemination strategy has successfully accelerated innovation in the transportation sector. NIST can also help lower risk associated with technology piloting by borrowing from the emerging piloting models in the water sector including the Isle Utilities “Trial Reservoirs” and WaterStart “CHANNELS” programs
Education, Workforce, and Community Engagement
Innovation in theory cannot become innovation in practice without a well-trained, certified workforce to implement new solutions. Now more than ever, Americans are in need of well-paying jobs. A new water workforce will provide job opportunities in every county in the nation. The next administration can begin retraining workers into tech-savvy water professionals immediately through programs like the National Science Foundation’s Advanced Technological Education program and the Department of Defense’s SkillBridge program.
Over the last three decades, the federal government has abdicated its responsibility for funding the water sector, leaving states and local utilities to tackle new treatment challenges, deal with the impacts of climate change, and overcome catastrophic events like the COVID-19 pandemic. The next Administration can reinvest in U.S. water systems and bolster our economy by empowering NIST to spearhead a Pipeline to deliver modern solutions for modern water obstacles.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
To meaningfully modernize U.S. water systems, the federal government should directly invest 1% of projected water infrastructure funding needs (about $12 billion over the next 10 years) into innovative water technology solutions. For comparison, recent investments into energy infrastructure such as DOE’s clean energy demonstrations have received $25 billion over the next 10 years. Federal spending on water infrastructure is nowhere near other peer infrastructure sectors and an order of magnitude lower than it is for the transportation sector.
The water sector is strongly influenced by federal regulations that maintain minimum drinking-water and wastewater treatment standards. Where existing treatment standards can be met with legacy technologies, there is little incentive for utilities to invest in newer, advanced systems. Furthermore, evaluation and approval of new technologies typically occurs on a state-by-state basis, where differing state regulatory requirements inhibit the dissemination of successful solutions across jurisdictional boundaries. These barriers mean that useful new technologies can take as long as a decade to see widespread deployment in the water sector.
Funding support allocated through the Pipeline will de-risk investment by individual water utilities and enable those utilities to adopt new technologies and upgrade to newer systems more easily and efficiently. The uptake of new technologies will in turn increase demand for new water innovations, creating market opportunities for U.S. start-ups and entrepreneurs. Overall, the Pipeline will increase public health protections through deployment of advanced treatment systems and monitoring solutions while also attracting additional private investment in the water sector.
The Pipeline will expedite development of new treatment technologies, monitoring systems, and cost-saving strategies, and will enable these solutions to be deployed at local utilities much more quickly. The Pipeline will support promising innovations and fund demonstration projects to increase the availability and visibility of vetted technology solutions for large and small utilities alike. Emphasis could and should be placed on accelerating technologies that solve challenges in rural communities, overburdened communities, communities with declining populations, and other underserved communities.
The Pipeline will require leadership and expertise from an agency focused on public-private partnerships and job creation. The DOC’s mission is “to promote job creation, economic growth, sustainable development, and improved living standards for all Americans by working in partnership with businesses, universities, communities, and workers.” NIST, within the DOC, is well positioned to support standards development, fund technology evaluations, support small manufacturers, and develop public-private partnership consortia. DOC would coordinate with EPA experts as needed to evaluate public health impacts and/or environmental compliance.
The Clean Water and Drinking Water State Revolving Funds (SRFs) are designed to provide some funding directly to states – and then subsequently to utilities – to support local infrastructure projects through loans and grants. This process is not well suited for the funding of demonstration projects nor rapid sharing of solutions both regionally and nationally. The purpose of the Pipeline, by contrast, would be to identify new technologies that solve immediate and emerging broad challenges facing utilities across the nation, and to directly fund projects or consortia. Technologies identified through the Pipeline could ultimately be incorporated into SRF projects where appropriate.
Promoting Fairness in Medical Innovation
There is a crisis within healthcare technology research and development, wherein certain groups due to their age, gender, or race and ethnicity are under-researched in preclinical studies, under-represented in clinical trials, misunderstood by clinical practitioners, and harmed by biased medical technology. These issues in turn contribute to costly disparities in healthcare outcomes, leading to losses of $93 billion a year in excess medical-care costs, $42 billion a year in lost productivity, and $175 billion a year due to premature deaths. With the rise of artificial intelligence (AI) in healthcare, there’s a risk of encoding and recreating existing biases at scale.
The next Administration and Congress must act to address bias in medical technology at the development, testing and regulation, and market-deployment and evaluation phases. This will require coordinated effort across multiple agencies. In the development phase, science funding agencies should enforce mandatory subgroup analysis for diverse populations, expand funding for under-resourced research areas, and deploy targeted market-shaping mechanisms to incentivize fair technology. In the testing and regulation phase, the FDA should raise the threshold for evaluation of medical technologies and algorithms and expand data-auditing processes. In the market-deployment and evaluation phases, infrastructure should be developed to perform impact assessments of deployed technologies and government procurement should incentivize technologies that improve health outcomes.
Challenge and Opportunity
Bias is regrettably endemic in medical innovation. Drugs are incorrectly dosed to people assigned female at birth due to historical exclusion of women from clinical trials. Medical algorithms make healthcare decisions based on biased health data, clinically disputed race-based corrections, and/or model choices that exacerbate healthcare disparities. Much medical equipment is not accessible, thus violating the Americans with Disabilities Act. And drugs, devices, and algorithms are not designed with the lifespan in mind, impacting both children and the elderly. Biased studies, technology, and equipment inevitably produce disparate outcomes in U.S. healthcare.
The problem of bias in medical innovation manifests in multiple ways: cutting across technological sectors in clinical trials, pervading the commercialization pipeline, and impeding equitable access to critical healthcare advances.
Bias in medical innovation starts with clinical research and trials
The 1993 National Institutes of Health (NIH) Revitalization Act required federally funded clinical studies to (i) include women and racial minorities as participants, and (ii) break down results by sex and race or ethnicity. As of 2019, the NIH also requires inclusion of participants across the lifespan, including children and older adults. Yet a 2019 study found that only 13.4% of NIH-funded trials performed the mandatory subgroup analysis, and challenges in meeting diversity targets continue into 2024 . Moreover, the increasing share of industry-funded studies are not subject to Revitalization Act mandates for subgroup analysis. These studies frequently fail to report differences in outcomes by patient population as a result. New requirements for Diversity Action Plans (DAPs), mandated under the 2023 Food and Drug Omnibus Reform Act, will ensure drug and device sponsors think about enrollment of diverse populations in clinical trials. Yet, the FDA can still approve drugs and devices that are not in compliance with their proposed DAPs, raising questions around weak enforcement.
The resulting disparities in clinical-trial representation are stark: African Americans represent 12% of the U.S. population but only 5% of clinical-trial participants, Hispanics make up 16% of the population but only 1% of clinical trial participants, and sex distribution in some trials is 67% male. Finally, many medical technologies approved prior to 1993 have not been reassessed for potential bias. One outcome of such inequitable representation is evident in drug dosing protocols: sex-aware prescribing guidelines exist for only a third of all drugs.
Bias in medical innovation is further perpetuated by weak regulation
Algorithms
Regulation of medical algorithms varies based on end application, as defined in the 21st Century Cures Act. Only algorithms that (i) acquire and analyze medical data and (ii) could have adverse outcomes are subject to FDA regulation. Thus, clinical decision-support software (CDS) is not regulated even though these technologies make important clinical decisions in 90% of U.S. hospitals. The FDA has taken steps to try and clarify what CDS must be considered a medical device, although these actions have been heavily criticized by industry. Finally, the lack of regulatory frameworks for generative AI tools is leading to proliferation without oversight.
Even when a medical algorithm is regulated, regulation may occur through relatively permissive de novo pathways and 510(k) pathways. A de novo pathway is used for novel devices determined to be low to moderate risk, and thus subject to a lower burden of proof with respect to safety and equity. A 510(k) pathway can be used to approve a medical device exhibiting “substantial equivalence” to a previously approved device, i.e., it has the same intended use and/or same technological features. Different technical features can be approved so long as there are no questions raised around safety and effectiveness.
Medical algorithms approved through de novo pathways can be used as predicates for approval of devices through 510(k) pathways. Moreover, a device approved through a 510(k) pathway can remain on the market even if its predicate device was recalled. Widespread use of 510(k) approval pathways has generated a “collapsing building” phenomenon, wherein many technologies currently in use are based on failed predecessors. Indeed, 97% of devices recalled between 2008 to 2017 were approved via 510(k) clearance.
While DAP implementation will likely improve these numbers, for the 692 AI-ML enabled medical devices, only 3.6% reported race or ethnicity, 18.4% reported age, and only .9% include any socioeconomic information. Further, less than half did detailed analysis of algorithmic performance and only 9% included information on post-market studies, raising the risk of algorithmic bias following approvals and broad commercialization.
Even more alarming is evidence showing that machine learning can further entrench medical inequities. Because machine learning medical algorithms are powered by data from past medical decision-making, which is rife with human error, these algorithms can perpetuate racial, gender, and economic bias. Even algorithms demonstrated to be ‘unbiased’ at the time of approval can evolve in biased ways over time, with little to no oversight from the FDA. As technological innovation progresses, especially generative AI tools, an intentional focus on this problem will be required.
Medical devices
Currently, the Medical Device User Fee Act requires the FDA to consider the least burdensome appropriate means for manufacturers to demonstrate the effectiveness of a medical device or to demonstrate a device’s substantial equivalence. This requirement was reinforced by the 21st Century Cures Act, which also designated a category for “breakthrough devices” subject to far less-stringent data requirements. Such legislation shifts the burden of clinical data collection to physicians and researchers, who might discover bias years after FDA approval. This legislation also makes it difficult to require assessments on the differential impacts of technology.
Like medical algorithms, many medical devices are approved through 510(k) exemptions or de novo pathways. The FDA has taken steps since 2018 to increase requirements for 510(k) approval and ensure that Class III (high-risk) medical devices are subject to rigorous pre-market approval, but problems posed by equivalence and limited diversity requirements remain.
Finally, while DAPs will be required for many devices seeking FDA approval, the recommended number of patients in device testing is shockingly low. For example, currently, only 10 people are required in a study of any new pulse oximeter’s efficacy and only 2 of those people need to be “darkly pigmented”. This requirement (i) does not have the statistical power necessary to detect differences between demographic groups, and (i) does not represent the composition of the U.S. population. The standard is currently under revision after immense external pressure. FDA-wide, there are no recommended guidelines for addressing human differences in device design, such as pigmentation, body size, age, and pre-existing conditions.
Pharmaceuticals
The 1993 Revitalization Act strictly governs clinical trials for pharmaceuticals and does not make recommendations for adequate sex or genetic diversity in preclinical research. The results are that a disproportionately high number of male animals are used in research and that only 5% of cell lines used for pharmaceutical research are of African descent. Programs like All of Us, an effort to build diverse health databases through data collection, are promising steps towards improving equity and representation in pharmaceutical research and development (R&D). But stronger enforcement is needed to ensure that preclinical data (which informs function in clinical trials) reflects the diversity of our nation.
Bias in medical innovation are not tracked post-regulatory approval
FDA-regulated medical technologies appear trustworthy to clinicians, where the approval signals safety and effectiveness. So, when errors or biases occur (if they are even noticed), the practitioner may blame the patient for their lifestyle rather than the technology used for assessment. This in turn leads to worse clinical outcomes as a result of the care received.
Bias in pulse oximetry is the perfect case study of a well-trusted technology leading to significant patient harm. During the COVID-19 pandemic, many clinicians and patients were using oximeter technology for the first time and were not trained to spot factors, like melanin in the skin, that cause inaccurate measurements and impact patient care. Issues were largely not attributed to the device. This then leads to underreporting of adverse events to the FDA — which is already a problem due to the voluntary nature of adverse-event reporting.
Even when problems are ultimately identified, the federal government is slow to respond. The pulse oximeter’s limitations in monitoring oxygenation levels across diverse skin tones was identified as early as the 1990s. 34 years later, despite repeated follow-up studies indicating biases, no manufacturer has incorporated skin-tone-adjusted calibration algorithms into pulse oximeters. It required the large Sjoding study, and the media coverage it garnered around delayed care and unnecessary deaths, for the FDA to issue a safety communication and begin reviewing the regulation.
Other areas of HHS are stepping up to address issues of bias in deployed technologies. A new ruling by the HHS Office of Civil Rights (OCR) on Section 1557 of the Affordable Care Act requires covered providers and institutions (i.e. any receiving federal funding) to identify their use of patient care decision support tools that directly measure race, color, national origin, sex, age, or disability, and to make reasonable efforts to mitigate the risk of discrimination from their use of these tools. Implementation of this rule will depend on OCR’s enforcement, and yet it provides another route to address bias in algorithmic tools.
Differential access to medical innovation is a form of bias
Americans face wildly different levels of access to new medical innovations. As many new innovations have high cost points, these drugs, devices, and algorithms exist outside the price range of many patients, smaller healthcare institutions and federally funded healthcare service providers, including the Veterans Health Administration, federally qualified health centers and the Indian Health Service. Emerging care-delivery strategies might not be covered by Medicare and Medicaid, meaning that patients insured by CMS cannot access the most cutting-edge treatments. Finally, the shift to digital health, spurred by COVID-19, has compromised access to healthcare in rural communities without reliable broadband access.
Finally, the Advanced Research Projects Agency for Health (ARPA-H) has a commitment to have all programs and projects consider equity in their design. To fulfill ARPA-H’s commitment, there is a need for action to ensure that medical technologies are developed fairly, tested with rigor, deployed safely, and made affordable and accessible to everyone.
Plan of Action
The next Administration should launch “Healthcare Innovation for All Americans” (HIAA), a whole of government initiative to improve health outcomes by ensuring Americans have access to bias-free medical technologies. Through a comprehensive approach that addresses bias in all medical technology sectors, at all stages of the commercialization pipeline, and in all geographies, the initiative will strive to ensure the medical-innovation ecosystem works for all. HIAA should be a joint mandate of Health and Human Services (HHS) and the Office of Science Technology and Policy (OSTP) to work with federal agencies on priorities of equity, non-discrimination per Section 1557 of the Affordable Care Act and increasing access to medical innovation, and initiative leadership should sit at both HHS and OSTP.
This initiative will require involvement of multiple federal agencies, as summarized in the table below. Additional detail is provided in the subsequent sections describing how the federal government can mitigate bias in the development phase; testing, regulation, and approval phases; and market deployment and evaluation phases.
Three guiding principles should underlie the initiative:
- Equity and non-discrimination should drive action. Actions should seek to improve the health of those who have been historically excluded from medical research and development. We should design standards that repair past exclusion and prevent future exclusion.
- Coordination and cooperation are necessary. The executive and legislative branches must collaborate to address the full scope of the problem of bias in medical technology, from federal processes to new regulations. Legislative leadership should task the Government Accountability Office (GAO) to engage in ongoing assessment of progress towards the goal of achieving bias-free and fair medical innovation.
- Transparent, evidence-based decision making is paramount. There is abundant peer-reviewed literature that examines bias in drugs, devices, and algorithms used in healthcare settings — this literature should form the basis of a non-discrimination approach to medical innovation. Gaps in evidence should be focused on through deployed research funding. Moreover, as algorithms become ubiquitous in medicine, every effort should be made to ensure that these algorithms are trained on representative data of those experiencing a given healthcare condition.
Addressing bias at the development phase
The following actions should be taken to address bias in medical technology at the innovation phase:
- Enforce parity in government-funded research. For clinical research, NIH should examine the widespread lack of adherence to regulations requiring that government funded clinical trials report sex, racial or ethnicity, and age breakdown of trial participants. Funding should be reevaluated for non-compliant trials. For preclinical research, NIH should require gender parity in animal models and representation of diverse cell lines used in federally funded studies.
- Deploy funding to address research gaps. Where data sources for historically marginalized people are lacking, such as for women’s cardiovascular health, NIH should deploy strategic, targeted funding programs to fill these knowledge gaps. This could build on efforts like the Initiative on Women’s Health Research. Increased funding should include resources for underrepresented groups to participate in research and clinical trials through building capacity in community organizations. Results should be added to a publicly available database so they can be accessed by designers of new technologies. Funding programs should also be created to fill gaps in technology, such as in diagnostics and treatments for high-prevalence and high-burden uterine diseases like endometriosis (found in 10% of reproductive-aged people with uteruses).
- Invest in research into healthcare algorithms and databases. Given the explosion of algorithms in healthcare decision-making, NIH and NSF should launch a new research program focused on the study, evaluation, and application of algorithms in healthcare delivery, and on how artificial intelligence and machine learning (AI/ML) can exacerbate healthcare inequities. The initial request for proposals should focus on design strategies for medical algorithms that mitigate bias from data or model choices.
- Task ARPA-H with developing metrics for equitable medical technology development. ARPA-H should prioritize developing a set of procedures and metrics for equitable development of medical technology. Once developed, these processes should be rapidly deployed across ARPA-H, as well as published for potential adoption by additional federal agencies, industry, and other stakeholders. ARPA-H could also collaborate with NIST on standards setting with NIST and ASTP on relevant standards setting. For instance, NIST has developed an AI Risk Management Framework and the ONC engages in setting standards that achieve equity by design. CMS could use resultant standards for Medicare and Medicaid reimbursements.
- Leverage procurement as a demand-signal for medical technologies that work for diverse populations. As the nation’s largest healthcare system, the Veterans Health Administration (VHA) can generate demand-signals for bias-free medical technologies through its procurement processes and market-shaping mechanisms. For example, the VA could put out a call for a pulse oximeter that works equally well across the entire range of human skin pigmentation and offer contracts for the winning technology.
Addressing bias at the testing, regulation, and approval phases
The following actions should be taken to address bias in medical innovation at the testing, regulation, and approval phases:
- Raise the threshold for FDA evaluation of devices and algorithms. Equivalency necessary to receive 510(k) clearance should be narrowed. For algorithms, this would involve consideration of whether the datasets for machine learning tactics used by the new device and its predicate are similar. For devices (including those that use algorithms), this would require tightening the definition of “same intended use” (currently defined as a technology having the same functionality as one previously approved by the FDA) as well as eliminating the approval of new devices with “different technological characteristics” (the application of one technology to a new area of treatment in which that technology is untested).
- Evaluate FDA’s guidance on specific technology groups for equity. Requirements for the safety of a given drug, medical device, or algorithm should have the statistical power necessary to detect differences between demographic groups and represent all end-users of the technology..
- Establish a data bank for auditing medical algorithms. The newly established Office of Digital Transformation within the FDA should create a “data bank” of healthcare images and datasets representative of the U.S. population, which could be done in partnership with the All of Us program. Medical technology developers could use the data bank to assess the performance of medical algorithms across patient populations. Regulators could use the data bank to ground claims made by those submitting a technology for FDA approval.
- Allow data submitted to the FDA to be examined by the broader scientific community. Currently, data submitted to the FDA as part of its regulatory-approval process is kept as a trade secret and not released pre-authorization to researchers. Releasing the data via an FDA-invited “peer review” step in the regulation of high-risk technologies, like automated decision-making algorithms, Class III medical devices, and drugs, will ensure that additional, external rigor is applied to the technologies that could cause the most harm due to potential biases.
- Establish an enforceable AI Bill of Rights. The federal government and Congress should create protections for necessary uses of artificial intelligence (AI) identified by OSTP. Federally funded healthcare centers, like facilities part of the Veterans Health Administration, could refuse to buy software or technology products that violate this “AI Bill of Rights” through changes to federal acquisition regulation (FAR).
Addressing bias at the market deployment and evaluation phases
- Strengthen reporting mechanisms at the FDA. Healthcare providers, who are often closest to the deployment of medical technologies, should be made mandatory reporters to the FDA of all witnessed adverse events related to bias in medical technology. In addition, the FDA should require the inclusion of unique device identifiers (UDIs) in adverse-response reporting. Using this data, Congress should create a national and publicly accessible registry that uses UDIs to track post-market medical outcomes and safety.
- Require impact assessments of deployed technologies. Congress must establish systems of accountability for medical technologies, like algorithms, that can evolve over time. Such work could be done by passing the Algorithmic Accountability Act which would require companies that create “high-risk automated decision systems” to conduct impact assessments reviewed by the FTC as frequently as necessary.
- Assess disparities in patient outcomes to direct technical auditing. AHRQ should be given the funding needed to fully investigate patient-outcome disparities that could be caused by biases in medical technology, such as its investigation into the impacts of healthcare algorithms on racial and ethnic disparities. The results of this research should be used to identify technologies that the FDA should audit post-market for efficacy or the FTC should investigate. CMS and its accrediting agencies can monitor these technologies and assess whether they should receive Medicare and Medicaid funding.
- Review reimbursement guidelines that are dependent on medical technologies with known bias. CMS should review its national coverage determinations for technologies, like pulse oximetry, that are known to perform differently across populations. For example, pulse oximeters can be used to determine home oxygen therapy provision, thus potentially excluding darkly-pigmented populations from receiving this benefit.
- Train physicians to identify bias in medical technologies and identify new areas of specialization. ED should work with medical schools to develop curricula training physicians to identify potential sources of bias in medical technologies and ensuring that physicians understand how to report adverse events to the FDA. In addition, ED should consider working with the American Medical Association to create new medical specialties that work at the intersection of technology and care delivery.
- Ensure that technologies developed by ARPA-H have an enforceable access plan. ARPA-H will produce cutting-edge technologies that must be made accessible to all Americans. ARPA-H should collaborate with the Center for Medicare and Medicaid Innovation to develop strategies for equitable delivery of these new technologies. A cost-effective deployment strategy must be identified to service federally-funded healthcare institutions like Veterans Health Administration hospitals and clinical, federally qualified health centers, and Indian Health Service.
- Create a fund to support digital health technology infrastructure in rural hospitals. To capitalize on the $65 billion expansion of broadband access allocated in the Bipartisan Infrastructure Bill, HRSA should deploy strategic funding to federally qualified health centers and rural health clinics to support digital health strategies — such as telehealth and mobile health monitoring — and patient education for technology adoption.
A comprehensive road map is needed
The GAO should conduct a comprehensive investigation of “black box” medical technologies utilizing algorithms that are not transparent to end users, medical providers, and patients. The investigation should inform a national strategic plan for equity and non-discrimination in medical innovation that relies heavily on algorithmic decision-making. The plan should include identification of noteworthy medical technologies leading to differential healthcare outcomes, creation of enforceable regulatory standards, development of new sources of research funding to address knowledge gaps, development of enforcement mechanisms for bias reporting, and ongoing assessment of equity goals.
Timeline for action
Realizing HIAA will require mobilization of federal funding, introduction of regulation and legislation, and coordination of stakeholders from federal agencies, industry, healthcare providers, and researchers around a common goal of mitigating bias in medical technology. Such an initiative will be a multi-year undertaking and require funding to enact R&D expenditures, expand data capacity, assess enforcement impacts, create educational materials, and deploy personnel to staff all the above.
Near-term steps that can be taken to launch HIAA include issuing a public request for information, gathering stakeholders, engaging the public and relevant communities in conversation, and preparing a report outlining the roadmap to accomplishing the policies outlined in this memo.
Conclusion
Medical innovation is central to the delivery of high-quality healthcare in the United States. Ensuring equitable healthcare for all Americans requires ensuring that medical innovation is equitable across all sectors, phases, and geographies. Through a bold and comprehensive initiative, the next Administration can ensure that our nation continues leading the world in medical innovation while crafting a future where healthcare delivery works for all.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
HIAA will be successful when medical policies, projects, and technologies yield equitable health care access, treatment, and outcomes. For instance, success would yield the following outcomes:
- Representation in preclinical and clinical research equivalent to the incidence of a studied condition in the general population.
- Research on a disease condition funded equally per affected patient.
- Existence of data for all populations facing a given disease condition.
- Medical algorithms that have equal efficacy across subgroup populations.
- Technologies that work equally well in testing as they do when deployed to the market.
- Healthcare technologies made available and affordable to all care facilities.
Regulation alone cannot close the disparity gap. There are notable gaps in preclinical and clinical research data for women, people of color, and other historically underrepresented groups that need to be filled. There are also historical biases encoded in AI/ML decision making algorithms that need to be studied and rectified. In addition, the FDA’s role is to serve as a safety check on new technologies — the agency has limited oversight over technologies once they are out on the market due to the voluntary nature of adverse reporting mechanisms. This means that agencies like the FTC and CMS need to be mobilized to audit high-risk technologies once they reach the market. Eliminating bias in medical technology is only possible through coordination and cooperation of federal agencies with each other as well as with partners in the medical device industry, the pharmaceutical industry, academic research, and medical care delivery.
A significant focus of the medical device and pharmaceutical industries is reducing the time to market for new medical devices and drugs. Imposing additional requirements for subgroup analysis and equitable use as part of the approval process could work against this objective. On the other hand, ensuring equitable use during the development and approval stages of commercialization will ultimately be less costly than dealing with a future recall or a loss of Medicare or Medicaid eligibility if discriminatory outcomes are discovered.
Healthcare disparities exist in every state in America and are costing billions a year in economic growth. Some of the most vulnerable people live in rural areas, where they are less likely to receive high-quality care because costs of new medical technologies are too high for the federally qualified health centers that serve one in five rural residents as well as rural hospitals. Furthermore, during continued use, a biased device creates adverse healthcare outcomes that cost taxpayers money. A technology functioning poorly due to bias can be expensive to replace. It is economically imperative to ensure technology works as expected, as it leads to more effective healthcare and thus healthier people.
Protecting U.S. Critical Infrastructure with Resilience Caches of Reusable Respirators
To help protect U.S. critical infrastructure workers from future pandemics and other biological threats, the next presidential administration should use the federal government’s grantmaking power to ensure ample supplies of high-quality respiratory personal protective equipment (PPE). The administration can take five concrete actions:
- The Office of Pandemic Preparedness and Response Policy (OPPR) can coordinate requirements for federal agencies and recipients of federal emergency/disaster preparedness funding to maintain access to at least one reusable respirator per critical employee.
- The Department of Labor’s Occupational Safety and Health Administration (OSHA) can initiate an occupational safety rule on reusable respirator resilience caches.
- The Department of Health and Human Services’ Administration for Strategic Preparedness and Response (ASPR) can require PPE manufacturers receiving federal funding to demonstrate their robustness to extreme pandemics.
- ASPR’s Strategic National Stockpile can start stockpiling reusable respirators.
- The Federal Emergency Management Agency (FEMA) can leverage its public outreach experience to increase “peacetime” adoption of reusable respirators.
These actions would complete the Biden Administration’s existing portfolio of efforts to reduce the likelihood of dangerous PPE shortages in the future, reaffirming executive commitment to protecting vulnerable workers, building a resilient national supply chain, and encouraging innovation.
Challenge and Opportunity
The next pandemic could strike at any time, and our PPE supply chain is not ready. Experts predict that the chance of a severe natural epidemic could perhaps triple in the next few decades, and advances in synthetic biology are increasing the risk of deliberate biological threats. As the world witnessed in 2020, disposable PPE can quickly become scarce in a crisis. Inadequate stockpiles left millions of workers with insufficient access to respiratory protection and often higher death rates than the general public—especially the critical infrastructure workers who operate the supply chains for our food, healthcare, public safety, and other essential goods and services. In future pandemics, which could have a 4% to 11%+ chance of occurring in the next 20 years based on historical extrapolations, PPE shortages could cause unnecessary infections, deaths, and burnout among critical infrastructure workers.
Figure 1. Notional figure from Blueprint Biosecurity’s Next-Gen PPE Blueprint demonstrating the need for stockpiling PPE in advance of future pandemics.
Recognizing the vulnerability of our PPE supply chain to future pandemics, Section 3.3 of the National Biodefense Strategy and Implementation Plan directs the federal government to:
Establish resilient and scalable supply and manufacturing capabilities for PPE in the United States that can: (a) enable a containment response for; and (b) meet U.S. peak projected demand for healthcare and other essential critical infrastructure workers during a nationally or internationally significant biological incident.
At a high level, securing the supply of PPE during crises is already understood as a national priority. However, despite the federal government’s past efforts to invest in domestic PPE manufacturing, production capacity will still take time to ramp up in future scenarios. Our current stockpiles aren’t large enough to bridge that gap. Some illustrative math: there are approximately 50 million essential workers in the United States, but as of 2022 our Strategic National Stockpile only had about 540 million disposable N95 respirators. This is barely enough to last 10 days, assuming each worker only uses one per day. (One per day is even a stretch: extended use and reuse of disposable N95s often leads to air leakage around wearers’ faces.) State- and local-level stockpiles may help, but many states have already started jettisoning their PPE stocks as purchases from 2020 expire and the prudence of paying for storage becomes less visible. PPE shortages may happen again.
Fortunately, there is existing technology that can reduce the likelihood of shortages while also protecting workers better and reducing costs: reusable respirators, like elastomeric half-mask respirators (EHMRs).
A single EHMR typically costs between $20 and $40. While the up-front cost of an EHMR is higher than the ~$1 cost of a disposable N95, a single EHMR can reliably last a worker for thousands of shifts over the entirety of a pandemic. Compared to disposable N95s, EHMRs are also better at protecting workers from infection, and workers prefer them to disposable N95s in risky environments. EHMR facepieces often have a 10-year shelf life, and filter cartridges typically have the same five-year shelf life of a typical disposable N95. A supply of EHMRs also takes up an estimated 1.5% of the warehouse space of the equivalent supply of disposable N95s.

Figure 2. The relative size of equivalent PPE stockpiles. Source: Blueprint Biosecurity’s Next-Gen PPE Blueprint.
Some previous drawbacks of EHMRs were their lack of filtration for exhaled air and the unclear efficacy of disinfecting them between uses. Both of those problems are on their way to being solved. The newest generation of EHMRs on the market (products like the Dentec Comfort-Air Nx and the ElastoMaskPro) provide filtration on both inhalation and exhalation, and initial results from ongoing studies presented by the National Institute for Occupational Safety and Health (NIOSH) have demonstrated that they can be safely disinfected. (Product links are for illustrative purposes, not endorsement.)
Establishing stable demand for the newest generation of EHMRs could drive additional innovation in product design or material use. This innovation could further reduce worker infection rates by eliminating the need for respirator fit testing, improving comfort and communication, and enabling self-disinfection. It could also increase the number of critical infrastructure workers coverable with a fixed stockpile budget by increasing shelf lives and reducing cost per unit. Making reusable respirators more protective, ergonomic, and storable would improve the number of lives they are able to save in future pandemics while lowering costs. For further information on EHMRs, the National Academies has published studies that explore the benefits of reusable respirators.
The next administration, led by the new OPPR can require critical infrastructure operators that receive federal emergency/disaster preparedness funding to maintain resilience caches of at least one reusable respirator per critical infrastructure worker in their workplaces—enough to protect those workers during future pandemics.
These resilience caches would have two key benefits:
- Because many U.S. critical infrastructure operators, from healthcare to electricity providers, receive federal emergency preparedness funds, these requirements would bolster our nation’s mission-critical functions against pandemics or other inhalation hazards like wildfire smoke. At the same time, the requirements would be tied to a source of funding that could be used to meet them.
- By creating large, sustainable private-sector demand for domestic respirators, these requirements would help substantially grow the domestic industrial base for PPE manufacturing, without relying on future warm-basing payments like those that Congress recently rescinded.
By taking action, the next administration has an opportunity to reduce the future burden on taxpayers and the federal government, help keep workers safe, and increase the robustness of domestic critical infrastructure.
Plan of Action
Recommendation 1. Require federal agencies and recipients of federal emergency/disaster preparedness funding to maintain access to at least one reusable respirator per critical employee.
OPPR can coordinate a process to define the minimum target product profile of reusable respirators that employers must procure. To incentivize continual respirator innovation, OPPR’s process can regularly raise the minimum performance standards of PPE in these resilience caches. These standards could be published alongside regular PPE demand forecasts. As products expire every 5 or 10 years, employers would be required to procure the new, higher standard.
OPPR can also convene representatives from each agency that administers emergency/disaster preparedness funding programs to critical infrastructure sectors and can align those agencies on language for:
- Requiring funding recipients to maintain access to resilience caches of enough PPE.
- Explicitly including “maintaining access to PPE resilience caches” as an acceptable and encouraged use of funding.
The Cybersecurity and Infrastructure Security Agency (CISA) within the Department of Homeland Security (DHS) can update its definition of essential workers and set guidelines for which employees would need a reusable respirator.
FEMA’s Office of National Continuity Programs can recommend reusable respirator stocks for critical staff at federal departments and agencies, and the Centers for Medicare and Medicaid Services (CMS) can also set a requirement for healthcare facilities as a condition of participation for receiving Medicare reimbursement.
Recommendation 2. Initiate an occupational safety rule on reusable respirator resilience caches.
To cover any critical infrastructure workplaces that are not affected by the requirements in Recommendation 1, OSHA can also require employers to maintain these resilience caches. This provision could be incorporated into a broader rule on pandemic preparedness, as a former OSHA director has suggested.
OSHA should also develop preemptive guidance on the scenarios in which it would likely relax its other rules. In normal times, employers are usually required to implement a full, costly Respiratory Protection Program (RPP) whenever they hand an employee an EHMR. An RPP typically includes complex, time-consuming steps like medical evaluations that may impede PPE access in crises. OSHA already has experience relaxing RPP rules in pandemics, and preemptive guidance on when those rules might be relaxed in the future would help employers better understand possible regulations around using their resilience caches.
Recommendation 3. Require PPE manufacturers receiving federal funding to demonstrate their robustness to extreme pandemics.
The DHS pandemic response plan notes that workplace absenteeism rates during extreme pandemics are projected to be to 40%.
U.S. PPE manufacturers supported by federal industrial base expansion programs, such as the investments managed by ASPR, should be required to demonstrate that they can remain operational in extreme conditions in order to continue receiving funding.
To demonstrate their pandemic preparedness, these manufacturers should have:
- An on-site stockpile of enough reusable respirators for the maximum number of workers they might hire during a manufacturing surge.
- A comprehensive pandemic response plan that addresses workplace transmission protection.
- Regular exercises and independent evaluations of their pandemic response plans.
Recommendation 4. Start stockpiling reusable respirators in the Strategic National Stockpile.
Inside ASPR, the Strategic National Stockpile should ensure that the majority of its new PPE purchases are for reusable respirators, not disposable N95s. The stockpile can also encourage further innovation by making advance market commitments for next-generation reusable respirators.
Recommendation 5. Leverage FEMA’s public outreach experience to increase “peacetime” adoption of reusable respirators.
To complement work on growing reusable respirator stockpiles and hardening manufacturing, FEMA can also help familiarize the workforce with these products in advance of a crisis. FEMA can use Ready.gov to encourage the general public to adopt reusable respirators in household emergency preparedness kits. It can also develop partnerships with professional groups like the American Industrial Hygiene Association (AIHA) or the Association for Health Care Resource & Materials Management (AHRMM) to introduce workers to reusable respirators and instruct them in their use cases during both business as usual and crises.
Conclusion
Given the high and growing risk of another pandemic, ensuring that we have an ample supply of highly protective respiratory PPE should be a national priority. With new reusable respirators hitting the market, the momentum around pandemic preparedness after the COVID-19 pandemic, and a clear opportunity to reaffirm prior commitments, the time is ripe for the next administration to make sure our workers are safe the next time a pandemic strikes.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
Yes. Throughout COVID-19, critical infrastructure worker unions have consistently advocated for EHMRs. Examples include the New York State Nurses Association (NYSNA) and the Service Employees International Union (SEIU) 121rn.
Unions have also been at the forefront of broader calls for securing PPE access in future pandemics; the California Nurses Association was the driving force behind California’s most recent PPE stockpiling laws.
Studies have also shown that workers prefer reusable respirators to disposable N95s in risky environments.
Not in the long run. As a single employee infection can cost $340 per day, it is more cost-effective for most employers to spend around $3 per critical employee per year for reusable respirators. For hospitals in states like California or New York, which mandate one- to three-month PPE stockpiles, switching those stockpiles to reusable respirators would likely be cost-saving, as demonstrated by past case studies. Most of these hospitals are still meeting those requirements with disposable N95s largely because of slow product choice re-evaluation cycles.
Managing these resilience caches would also pose a minimal burden on employers. Most EHMRs can be comfortably stored in most indoor workplaces, taking up around the volume of a large coffee mug for each employee. Small workplaces with fewer than 50 employees could likely fit their entire resilience cache in a cardboard box in a back closet, and large workplaces will likely already have systems for managing emergency products that expire, like AEDs, first-aid kits, and fire extinguishers. As with other consumables like printer ink cartridges, PPE manufacturers can send reminders to employers when the products they purchased are about to expire.
To put this into perspective, fire alarm units should generally be replaced every 10 years for $20 to $30 each, and typically require new batteries once or twice per year. We readily accept the burden and minor cost of fire alarm maintenance, even though all U.S. fire deaths in the last 10 years only accumulate to 3% of COVID-19’s U.S. death toll.
While EHMRs fit most workers, there may be some workers who aren’t able to wear them due to religious norms or assistive devices.
Those workers can instead wear another type of reusable respirator, powered air-purifying respirators (PAPRs). While PAPRs are even more effective at keeping workers safe than EHMRs, they cost significantly more and can be very loud. Employers and government stockpiles can include a small amount of PAPRs for those workers who can’t wear EHMRs, and can encourage eventual cost reductions and user-experience improvements with advance market commitments and incremental increases in procurement standards.
Yes, but they reduce the risk of any lags in PPE access. Every day that workers are exposed to pathogens without adequate PPE, their likelihood of infection goes up. Any unnecessary exposures speed the spread of the pandemic. Also, PPE manufacturing ramp-up could be slowed by employee absences due to infection or caring for infected loved ones.
To accommodate some employers’ reluctance to build physical stockpiles, the administration can enable employers to satisfy the resilience cache requirement in multiple ways, such as:
- On-site resilience caches in their workplaces
- Agreements with distributors to manage resilience cache inventory as a rotating supply bubble
- Agreements with third-party resilience cache managers
- Purchase options with manufacturers that have demonstrated enough capacity to rapidly manufacture resilience cache inventory at the start of a pandemic
Purchase options would function like a “virtual” resilience cache: they would incentivize manufacturers to build extra warm-base surge capacity and test their ability to rapidly ramp up manufacturing pace. However, it would increase the risk that workers will be exposed to infectious disease hazards before their PPE arrives. (Especially in the case of a severe pandemic, where logistics systems could get disrupted.)
Yes. Employers could use respirators from their resilience cache to protect workers from localized incidents like seasonal flu outbreaks, wildfires, or smog days, and put them back into storage when they’re no longer needed.
Yes. Federal emergency/disaster preparedness funding could be tied to other requirements, like:
- Installing the capacity to turn up workplace air ventilation or filtration significantly
- Maintaining and regularly exercising pandemic response protocols
- Investing in passive transmission suppression technology (e.g., germicidal ultraviolet light)
The next time there’s a pandemic, having these requirements in place could help ensure that any post-emergency funding (e.g., Hazard Mitigation Assistance Program grants) will be spent on innovative PPE that aligns with the federal government’s broader PPE supply chain strategies.
If the Strategic National Stockpile receives additional post-emergency funding from Congress, it could also align its purchases with the target product profiles that critical infrastructure operators are already procuring to.
A Guide to Public Deliberation
Science is advancing at an unprecedented speed, and scientists are facing major ethical dilemmas daily. Unfortunately, the general public rarely gets opportunities to share their opinions and thoughts on these ethical challenges, moving us, as a society, towards a future that is not inclusive of most people’s ideas and beliefs. Scientists regularly call for public engagement opportunities to discuss cutting-edge research. In fact, “71% of scientists [associated with the American Association for the Advancement of Science (AAAS)] believe the public has either some or a lot of interest in their specialty area.” Sadly, scientists’ calls often go unnoticed and unanswered, as there continue to be inadequate mechanisms for these engagement opportunities to come to fruition.
To Deliberate or Not to Deliberate
Public deliberation, when performed well, can lead to more transparency, accountability to the public, and the emergence of ideas that would otherwise go unnoticed. Due to the direct involvement of participants from the public, decisions made through such initiatives can also be seen as more legitimate. On a societal level, public deliberation has been shown to encourage pluralism among participants.
Despite the importance of deliberation, it’s important to note that it is not always the best way to engage the public. Planning a public deliberation event — a citizens’ panel, for instance — takes a large amount of time and resources. Plus, incentivizing a random sample of citizens to participate (which is considered the gold standard of deliberation) is difficult. It’s therefore paramount to first assess whether the topic of focus is suitable for public deliberation.
To assess the appropriateness of a deliberation topic, consider the following criteria (inspired by criteria set forth by Stephanie Solomon and Julia Abelson and the Kettering Foundation):
- Does the issue involve conflicting public opinions? Issues that involve setting priorities in healthcare, for example, may benefit from public deliberation as there is no singular correct answer; deliberation may offer a more clear and holistic view of what is best for a community, according to the community.
- Is the issue controversial? If so, deliberation can be a good tool as it brings many opinions into view and can foster pluralism as mentioned previously.
- Does the issue have no clear-cut solution and is “intractable, ongoing, or systemic”?
- Do all available solutions have significant drawbacks?
- Does the community at large have an interest in the problem?
- Would the discussion of the issue benefit from a combination of expert and real-world experience and knowledge (what Solomon and Abelson call “hybrid” topics)? Certain issues may solely require technical knowledge but many issues would benefit from the views of the public as well.1
- Are citizens and the government on the same page about the issue? If not, public deliberation can foster trust, but only if the initiative is done with the intention of taking the public’s conclusions into account.
Setting Goals
If it’s deemed that the topic is suitable for public deliberation, the next step is to set goals for the public deliberation initiative. Julia Abelson, Lead of the Public Engagement in Health Policy Project and Professor at McMaster University, has explained that one of the significant differentiating factors between successful and unsuccessful initiatives is thoughtful planning and organization — including setting clear goals and objectives organizers would like to meet by the end of deliberation. Having an end goal not only helps with planning but also allows for a realistic goal to be shared with deliberation participants. Setting unrealistic expectations as to what the deliberation process is meant to achieve — and subsequently not achieving those goals — will lead participants and citizens, in general, to lose trust in the deliberation process (and organizational body).
Is the goal of deliberation to bring new ideas into view and share those with relevant agencies (governmental or otherwise)? Is the goal instead to enact change in current policies? Is the goal to help shape new policies? The aforementioned Citizens’ Reference Panel on Health Technologies in Canada did not directly impact the government’s decisions, but served to make experts aware of a viewpoint they had not previously explored. This is in contrast to the typical “sit and listen” initiatives that don’t have as much of a capacity to encourage new ideas to emerge. In another instance, a citizens’ jury in Buckinghamshire, England was formed to discuss how to tackle back pain in the county. The Buckinghamshire Health Authority promised to implement the citizens’ recommendations (as was mandated by a charity that was supporting this public deliberation effort) — and they did.
Expanding on the idea of making promises and accountability, it’s important for the organizing body — which may or may not include a federal agency — to consider its role in implementing the conclusions of the deliberation. Promising to implement the conclusion of the deliberations can serve to invigorate discussion and make participants more engaged, knowing that their discussions can have a direct impact on future decisions. For instance, the British Columbia Biobank Deliberation involved a “commitment at the outset of the deliberation from the leaders of a proposed BC BioLibrary (now funded by the Michael Smith Foundation for Health Research) that the Bio-Library’s policy discussions would consider suggestions from this deliberation.” Researchers have suggested this may have contributed to participants’ interest in the deliberation event. Despite some examples of implementation following deliberation (such as the Buckinghamshire and Ontario examples), there continues to be a lack of adequate change based on the public’s recommendations. One other instance comes from NASA’s 2014 efforts to involve the public in the discussion around planetary defense (in the context of asteroids) through a participatory technology assessment (PTA). It seems that the PTA helped to spur the creation of NASA’s Planetary Defense Coordination Office.
Furthermore, providing updates on implementation to participants, and the public at large, would provide another crucial aspect of accountability: “explanations and justifications.” However, these updates on their own would not fulfill an organization or agency’s duty to accountability as that requires an active dialogue with the public (which is precisely why implementing the conclusions of public deliberation initiatives is important).
When to Deliberate: Agenda Setting for Citizens
As mentioned above, deliberation can happen at various points during the policymaking pipeline. It has become increasingly popular to include the public early on in the process, such as in an agenda-setting role. This allows the public not only to engage in discussions about a topic but to also set the priorities and frame how the discussions will move forward. As Naomi Scheinerman writes, “with proper agenda setting and precedent creation, the resulting […] questions would be more reflective of what the public is interested in discussing rather than of the companies, industries, and other stakeholder groups.”
A trailblazing model in citizen agenda-setting has been the Ostbelgien Model. The model involves both a permanent Citizens’ Council and ad hoc Citizens’ Panels. Though the members of the Citizens’ Council rotate (and are chosen randomly), one of the permanent roles of the Council is to select topics for the ad hoc Citizens’ Panels, with citizens having a direct hand in what issues their fellow citizens and government should tackle. Since its inception in 2019, the Citizens’ Council has asked Citizens’ Panels to tackle issues such as “how to improve the working conditions of healthcare workers” and “inclusive education.”
Framing
One of the pillars of the success of public deliberation is a well-scoped question that is framed appropriately. Issues that are framed unfairly, meaning they place emphasis on a specific part of the issue while ignoring others, can lead to inaccurate results and a loss of trust between the public and the organizers. Though this depends on the goals of the deliberation, it’s often best for questions to be specific in their scope to allow for concrete results at the end of the deliberation initiative. For example, an online deliberation session in New York City aimed to assess the public’s views on who should be given priority access to COVID-19 vaccines. One of the questions asked participants to rank the order in which they think a pre-specified list of essential workers should get access to the vaccine. This allows for discussion while retaining a clear focus.
Another example comes from climate change. Climate change can be framed in many ways — through an economic frame, a public health frame, a justice frame, and others. These various framings impact how the public reacts to the issue; in the case of the economic frame, it has led to “political divisiveness.” Focusing instead on the public health frame, for instance, led to greater agreement on policy decisions. Similarly, according to a 2023 policy paper from the Organisation for Economic Co-operation and Development (OECD), an issue like COVID-19 can be less polarizing if the framing used is about solutions to the pandemic rather than solely vaccines. Importantly, the organizers of the public deliberation initiative do not have sole control over the framing of the issue. Citizens often have a pre-existing “frame of thought.” This makes frames tricky yet essential in making it possible to appropriately and productively deliberate a topic.
Framing is implicit in that participants in deliberation are not aware of it, making it all the more crucial to be wary of the framing. Thus, it becomes clear how seemingly unimportant factors, such as setting, also affect deliberation. According to Mauro Barisione, the framing of the setting includes:
- Who is promoting the event and who the sponsors are: Whether the public trusts the promoters/sponsors and the feelings they associate with them (including any explicit views the sponsors promote) may impact the framing of the topic at hand.
- Where is/are the deliberative session(s) being held “(i.e., institutional, academic, civil society or business organization, etc.)”: Apart from accessibility concerns, the location of the deliberative session(s) sends implicit messages to participants who may have previously had negative experiences at governmental institutions, for instance.
- Who are the witnesses and experts brought on as part of the project: This point is most obvious. Especially when it comes to less well-known topics, witnesses and experts may be the first window into a new topic for participants. A biased selection of experts (or including experts who provide unscientific evidence) can significantly impact deliberation. Moreover, an oft-neglected point worth considering is the diversity of expert witnesses who present relevant information at public deliberation events. Selecting experts is discussed in more detail in a later section, but it’s worth noting here that a diverse group of expert presenters will ensure a diversity of views and may aid in building trust with participants as well.
Selecting a Type of Public Deliberation
Another factor that merits attention at this point is the type of public deliberation being undertaken. Though public deliberation has been referred to as one entity thus far, there are many different types, including, but not limited to, citizens’ juries, planning cells, consensus conferences, citizens’ assemblies, and deliberative polls. Below are some further details about various types of public deliberation (where a source is not included below, it was adapted from Smith & Setälä).
Citizens’ juries
- Description: “In common with the legal jury, citizens’ jury assumes that a small group of ordinary people, without special training, is willing and able to take important decisions in the public interest” (Coote & Mattinson)
- Selected jurists are meant to represent a microcosm of their community (Crosby)
- Jurors are selected through a quota process that often takes into account demographics (e.g., age, gender, education and race) or jurors’ prior opinions about the question at hand (Crosby)
- Neutral moderator moderates discussions (Crosby)
- Jurors are able to determine what questions are asked of the witnesses
- Similar to planning cells (below), jurors are paid for the time (e.g., $75 per day)
- Number of participants: 12–24
- Time: 2-4 days (Armour)
- Output: Jurors put together a written report of their conclusions (Armour)
- Examples: Ned Crosby of the independent Jefferson Institute in the US initially ran them with groups of 12-24 people (Smith & Wales)
- Another example is the Oregon Citizens’ Initiative Review
Planning cells
- Description: Deliberation includes 3 phases (Participedia):
- Citizens are presented with pertinent information from multiple perspectives and ask clarifying questions
- Cells are broken up into smaller groups of 5 and work together to come up with recommendations regarding the topic
- The full cell reconvenes and each mini group presents their top recommendation, which is then assessed by all members of the group, until some final recommendations are made
- Number of participants: 25 in each cell (multiple cells running simultaneously or one after another); often have 6-10 planning cells total (Dienel)
- Time: 3-4 days (Dienel)
- Each day is split into four chunks of time, each spent on a different “thematic focus”
- The final day is used to summarize the participants’ thoughts and come to conclusions (Planning Cells Database)
- Output: Citizens’ report
- Created by the planning committee based on quantitative data aggregated from all the cells (for instance, from how participants responded to which option they favored) (Dienel)
- The first draft of the report may also be discussed with participants in a follow-up meeting
- Example: Cologne Town Square
Consensus conferences/citizens’ conferences
- Description: These conferences have been described as a citizens’ jury plus a town hall meeting (Einsiedel & Eastlick)
- Citizens learn some basic facts about the issue at hand and formulate questions they’d like addressed (Kenyon). In other words, the topic is chosen by the organizers while the concrete problems are selected by the citizens (Nielsen et al.).
- Witnesses are called to answer these questions (Kenyon) and the experts are selected entirely by citizens (Nielsen et al.)
- A steering group is often involved who ensures that “the conference is balanced and just” and provides support to the organizers (Nielsen et al.)
- Members include “scientists, representatives of non-governmental organizations, policy-makers and others, who are engaged and informed concerning the topic”
- It’s recommended that fact sheets be prepared that explain basic definitions necessary for understanding the topic as well as pros and cons for all sides — this will be provided to participants in preparation for discussions and the questioning of expert witnesses (Nielsen et al.)
- Introductory materials should also clearly explain the role of the consensus conference participants and how their contributions will be used to inform future decisions
- Number of participants: 10-24
- Time: 4 days
- Output: Report written by citizen participants (Kenyon)
- Example: Danish Board of Technology consensus conferences
Citizens’ assemblies
- Description: Typically larger and longer than the deliberative processes discussed above, similar to legislatures composed of regular citizens (Lacelle-Webster & Warren)
- Organizers often aim to make assemblies as representative of the public as possible through methods like stratified random sampling (Lacelle-Webster & Warren)
- More limited participation opportunities so some suggest that it’s better to call citizens’ assemblies “representative institutions” rather than a form of participatory democracy (Lacelle-Webster & Warren)
- Participants compensated for their time (Ferejohn)
- Based on the initial citizens’ assembly in British Columbia, any interested expert witnesses or groups were able to testify in front of the participants (Ferejohn)
- Participants can ask any questions of the witnesses (Ferejohn)
- A Chair — who was the only public official directly involved in the process — is able to make obligatory rulings as to how the deliberations would proceed
- Organizers set the agenda
- Formats can vary: the inaugural 2004 British Columbia citizens’ assembly was more so in the format of a legislature as opposed to an Irish citizens’ assembly which included roundtable discussions with groups of 7-8 participants alongside a facilitator and notetaker at each table (Farrell et al.)
- Number of participants: 99-150
- Time: Meetings over several months (often during weekends) (Farrell et al.)
- Output: Report and recommendations from citizens’ assembly (Ferejohn)
- Example: The 2004 British Columbia Citizens’ Assembly
Deliberative polls
- Description: Combines traditional polling with the benefits of participatory processes
- Randomly selected participants are polled on their opinions about a topic (Stanford Deliberative Democracy Lab)
- Participants receive introductory materials about the issue prior to the event (Stanford Deliberative Democracy Lab)
- A weekend-long event where participants learn more about the issues from experts (ask questions from experts) and discuss issues with one another with trained facilitators present (Stanford Deliberative Democracy Lab)
- At the end, complete the questionnaire from earlier once more (Stanford Deliberative Democracy Lab)
- Participants often paid for their time ($75-$200) (Participedia)
- Number of participants: 200+
- Time: One weekend (Stanford Deliberative Democracy Lab)
- Output: Differences measured between the opinions of participants pre- and post-deliberative polling process (Stanford Deliberative Democracy Lab)
- Example: 2008 deliberative poll on housing shortages in San Mateo County, California
A note on online deliberation
The COVID-19 pandemic forced many initiatives to shift to a fully online modality. This highlighted many of the opportunities as well as challenges that online deliberation presents. One consideration is accessibility, a double-edged sword when it comes to deliberation. Virtual deliberation alleviates the need for a venue or hotel accommodations — decreasing costs for organizers — and may allow participants to continue to go to work at the same time. However, difficulties with using technology and a lack of access to a device or an internet connection are drawbacks. Another opportunity presented by virtual deliberation is to provide more balanced viewpoints on the topic of deliberation. For instance, there are no geographical barriers as to the experts organizers can invite to speak at an event.
A concern somewhat unique to online deliberation is data privacy and security. While this can also be an issue with in-person initiatives, many tools that participants are familiar with and may prefer to use do not have robust security.
A note on cost
While the cost of many deliberation initiatives is not publicly available, the available estimates range from $20,000 (citizens’ jury) to $95,000 (consensus conference) to $2.6 million (Europe-wide deliberative poll of 4300 people) to $5.5 million (citizens’ assembly). Note that these costs come from a range of time points and locations (though they have been adjusted for inflation) and only serve as rough estimates. A major contributor to these costs, particularly for longer deliberative initiatives, is hotel or venue costs as well as the reimbursement of participants. This reimbursement is costly but a part of the founding philosophy of many types of deliberation, including that of planning cells.
Selecting Participants
Many different approaches can be taken to selecting participants for deliberative forums. Unfortunately, there are inherent trade-offs in selecting a sampling method or approach. For instance, random sampling is more in line with the principle of “equal opportunity” and may promote “cognitive diversity”— the diversity of ideas, experiences, and approaches participants bring to the event — but is prone to creating deliberation groups that are not representative of the population at large. This is particularly true when the deliberative forum has few participants. This is why, depending on the type of deliberation event (and therefore number of participants chosen), a different type of sampling may be appropriate.
Another approach is random-stratified sampling, where participants are randomly chosen and invited to participate in the deliberative event. There is often an unequal distribution among those who accept the invitation — for instance, individuals with higher socio-economic statuses may respond disproportionately more. In this case, a more representative sample may be chosen from those who responded. Quotas may also be set, such as ensuring that a certain number of female-identifying participants are included in a deliberative event. For this method, the organizers must decide on groups of individuals who are primarily affected by the topic being discussed, as well as groups often excluded from such deliberations. A deliberative forum on immigration, for instance, may call for the presence of a participant who is an immigrant to ensure polarization does not take place. In certain instances, purposive sampling — where individuals from groups whose views are specifically being sought are purposefully chosen — may also be appropriate. Furthermore, some researchers suggest including a “critical mass” of individuals from typically underserved groups. This can serve to make participants more comfortable in speaking up, ensure that the diversity of discussions is retained when participants are broken up into smaller groups (in certain forms of public deliberation), and provide a step in avoiding tokenism.
Furthermore, there are newer methods of selecting participants that combine both random and stratified sampling — namely algorithms that try to maximize both representation and equal opportunity of participation. One instance is the LEXIMIN algorithm which “choose[s] representative panels while selecting individuals with probabilities as close to equal as mathematically possible.” This algorithm is open-access and can be used at panelot.org.
Aside from considerations for selecting participants, it’s important to consider the selected individuals’ ability and willingness to participate. Several factors can dissuade selected individuals from taking part, including but not limited to, the cost of missing work, the cost of childcare, transportation costs, and lack of trust in the organizing body or agency. Prohibitive costs are addressed by several of the deliberation models discussed in the “Selecting a Type of Public Deliberation” section. These models strongly suggest stipends which, at minimum, cover incidental expenses. A lack of trust is a particularly important issue to address as it can hinder the organizer’s ability to reach individuals typically left out of policymaking discussions. One approach to addressing this once again brings us to making — and critically, keeping — promises regarding the implementation of the conclusions of participants. Framing (as discussed in an earlier section) can also contribute to building trust, though, importantly, this is not a gap that can be bridged overnight. A more extensive discussion on inclusion in public deliberation forums can be found here.
Bringing On Experts & Creating Materials
Prior to selecting the group who will participate in the public deliberation activity, steps need to be taken to organize which experts will be part of the event and create the informational material that will be provided to participants before deliberations begin.
Here, efforts must be made to ensure sufficient and balanced information is presented without creating a framing event where participants enter discussions with a biased perspective. It has been found that participants readily integrate the facts and opinions presented by experts/witnesses prior to deliberation and critically engage with their points. A deliberative engagement initiative in British Columbia, Canada about biobanking brought on a variety of experts and stakeholders to present to participants. To ensure fairness, presenters were “given specific topics, limited presentation times, and asked to use terms as defined in the information booklet” that was previously provided. A unique component included in this initiative was the ability for participants to ask presenters questions in between the two deliberative session weekends, which were two weeks apart, through a website.
In addition, participants were provided with booklets and readings. In the case of the British Columbia initiative, to create booklets and background materials, a literature review was performed. Once more, the materials should provide a balance of opinions. They should include the most important facts relevant to the question at hand, some of the most common/salient approaches and points with regards to the question, and the weaknesses of each approach/point (Mauro Barisione). It is also best to keep materials succinct, with some deliberative initiatives keeping their materials to one page long.
Though the traditional approach is to have experts present prior to deliberation, other methods have also been used. For instance, a Colorado deliberation initiative focused on future water supply used an “on tap but not on top” expert approach. Rather than call experts to present information, they instead provided one-page information sheets, followed directly by deliberation. Experts were present during the deliberation session. When prompted by a participant, a facilitator would ask an expert to briefly join the group to answer the participant’s question. The approach was largely successful, though one “rogue expert” frequently interjected in a group’s discussion, providing his own opinions. One limiting factor to this approach is time; the deliberative sessions mentioned above were two hours long. But many other forms of deliberation are significantly longer, making coordinating with experts for long durations of time difficult. Despite these challenges, this approach provides an interesting way of integrating experts into the deliberation process so their expertise is best used and the participants’ questions are best answered as they arise.
Facilitation
A good facilitator or moderator is critical to the deliberation process. As explained by Kara N. Dillard, moderators set the ground rules for the discussion and prevent any one participant from dominating the session; this is called presentation. It has been found that clearly setting expectations for the discussion can lead to greater deliberative functioning — which, for our purposes, includes the exchange of ideas/reasons, equality, and freedom to speak and be heard — according to participants. Moderators also guide the discussion in two main ways: asking questions that challenge what participants have already discussed (elicitation); and connecting ideas that were previously brought up to new topics and “play[ing] devil’s advocate” to bring forth new ideas (interpretation). At the end of the session, moderators also help participants produce conclusions by asking what areas of consensus and contention were present throughout the discussion.
Moderators can take multiple approaches to facilitating, with one framework proposed by Kara N. Dillard separating moderators into three groups: passive, moderate, and involved. Passive moderators take a “backseat” approach to moderating. They often describe their role to participants as only being there to prevent a participant from dominating the conversation, rather than actively leading it. This has led to unfocused discussions and unclear conclusions. Participants often jumped around and went off-topic. Though this passive approach may work in some instances, a moderate or involved approach often leads to better deliberation.
Involved facilitators actively lead the discussion by asking questions that challenge participants to think in new ways, sometimes acting as a “quasi-participant.” In line with this, these moderators often play devil’s advocate to move the discussion in new, albeit related, directions. These moderators ask follow-up questions and “editorialize” to help participants flesh out their ideas together and aim to pinpoint points of contention so participants can further discuss them. If participants begin to veer off-topic, involved moderators will move the group back into a more focused direction while also connecting this new topic to the main question, allowing for new thoughts to emerge. These moderators take the time to sum up the main points brought up by participants after each point so conclusions become clear. Once more, this approach may not work in all instances but often leads to deeper conversations and more focused conclusions.
As implied by the name, moderate facilitators are somewhere in between passive and involved facilitators. These moderators ask questions to guide the discussion, but don’t often challenge the participants and let them take the wheel. These moderators use the elicitation strategy frequently, an important difference between moderate and passive moderators.
Due to the skills needed to facilitate a deliberation event well, organizers or government agencies looking to organize these events may require would-be facilitators to undergo brief training.
What Comes Next
After deliberation has taken place, the next step is to write a report summarizing the conclusions of the deliberative forum. As we have seen several times with other topics, there are multiple approaches to this. One approach is to leave the report writing to the facilitators, organizers, or researchers who use their own takeaways from the deliberation (in the case of facilitators) or summarize based on recordings or transcripts (in the case of organizers or researchers). However, this method introduces bias into the process and doesn’t allow participants to be directly involved in creating conclusions or next steps.
An alternative is to allocate time towards coming up with conclusions together with participants both throughout and at the end of the deliberative session. Recall that involved facilitators frequently summarize the conclusions of the group throughout the deliberation, making this final task both more efficient and more participant-led. Participants can directly and immediately add on to or push back against the facilitator’s summary. As a guideline, Public Agenda, an organization conducting public engagement research, divides the summary into the following sections: areas of agreement, areas of disagreement, questions requiring further research, and high-priority action steps.
A Dose Of Reality: Underscoring The Fatal Consequences Of The Opioid Epidemic
The opioid epidemic is a public health and safety emergency that is killing thousands and destroying the quality of life for hundreds of thousands of Americans and those who care about them. Fentanyl and other opioids affect all age ranges, ethnicities, and communities, including our most vulnerable population, children. Producing fentanyl is increasingly cheap, costing pennies for a fatal dose, with the opioid intentionally or unintentionally mixed with common illicit street drugs and pressed into counterfeit pills. Fentanyl is odorless and tasteless, making it nearly untraceable when mixed with other drugs. Extremely small doses of fentanyl, roughly equivalent to a few grains of salt, can be fatal, while carfentanil, a large animal tranquilizer, is 100 times more potent than fentanyl and fatal at an even smaller amount.
The Biden-Harris Administration should do even more to fund opioid-related prevention, treatment, eradication, and interdiction efforts to save lives in the United States. The 2022 Executive Order to Address the Opioid Epidemic and Support Recovery awarded $1.5 billion to states and territories to expand treatment access, enhance services in rural communities, and fund law enforcement efforts. In his 2023 State of the Union address, President Biden highlighted reducing opioid overdoses as part of his bipartisan Unity Agenda, pledging to disrupt trafficking and sales of fentanyl and focus on prevention and harm reduction. Despite extensive funding, opioid-related overdoses have not significantly decreased, showing that a different strategy is needed to save lives.
Opioid-related deaths have been estimated cost the U.S. nearly $4 trillion over the past seven years—not including the human aspect of the deaths. The cost of fatal overdoses was determined to be $550 billion in 2017. The cost of the opioid epidemic in 2020 alone was an estimated $1.5 trillion, up 37% from 2017. About two-thirds of the cost was due to the value of lives lost and opioid use disorder, with $35 billion spent on healthcare and opioid-related treatments and about $15 billion spent on criminal justice involvement. In 2017, per capita costs of opioid use disorder and opioid toxicity-related deaths were as high as $7247, with the cost per case of opioid use disorder over $221,000. With inflation in November 2023 at $1.26 compared to $1 in 2017, not including increases in healthcare costs and the significant increase in drug toxicity-related deaths, the total rate of $693 billion is likely significantly understated for fatal overdoses in 2023. Even with extensive funding, opioid-related deaths continue to rise.
With fatal opioid-related deaths being underreported, the Centers for Disease Control and Prevention (CDC) must take a primary role in real-time surveillance of opioid-related fatal and non-fatal overdoses by funding expanded toxicology testing, training first responder and medicolegal professionals, and ensuring compliance with data submission. The Department of Justice (DOJ) should support enforcement efforts to reduce drug toxicity-related morbidity and mortality, with the Department of Homeland Security (DHS) and the Department of the Treasury (TREAS) assisting with enforcement and sanctions, to prevent future overdoses. Key recommendations for reducing opioid-related morbidity and mortality include:
- Funding research to determine the efficacy of current efforts in opioid misuse reduction and prevention.
- Modernizing data systems and surveillance to provide real-time information.
- Increasing overdose awareness, prevention education, and availability of naloxone.
- Improve training of first responders and medicolegal death investigators.
- Funding rapid and thorough toxicology testing in emergency departments and coroner/medical examiner agencies.
- Enhancing prevention and enforcement efforts.
Challenge and Opportunity
Opioids are a class of drugs, including pain relievers that can be illegally prescribed and the illicit drug heroin. There are three defined waves of the opioid crisis, starting in the early 1990s as physicians increasingly prescribed opioids for pain control. The uptick in prescriptions stemmed from pharmaceutical companies promising physicians that these medications had low addiction rates and medical professionals adding pain levels being added to objective vital signs for treatment. From 1999 to 2010, prescription opioid sales quadrupled—and opioid-related deaths doubled. During this time frame when the relationship between drug abuse and misuse was linked to opioids, a significant push was made to limit physicians from prescribing opioids. This contributed to the second wave of the epidemic, when heroin abuse increased as former opioid patients sought relief. Heroin-related deaths increased 286% from 2002 to 2013, with about 80% of heroin users acknowledging that they misused prescription opioids before using heroin. The third wave of the opioid crisis came in 2013 with an increase in illegally manufactured fentanyl, a synthetic opioid used to treat severe pain that is up to 100 times stronger than morphine, and carfentanil, which is 100 times more potent than fentanyl.
In 2022, nearly 110,000 people in the United States died from drug toxicity, with about 75% of the deaths involving opioids. In 2021, six times as many people died from drug overdoses as in 1999, with a 16% increase from 2020 to 2021 alone. While heroin-related deaths decreased by over 30% from 2020 to 2021, opioid-related deaths increased by 15%, with synthetic opioid-involved deaths like fentanyl increasing by over 22%. Over 700,000 people have died of opioid-related drug toxicity since 1999, and since 2021 45 people have died every day from a prescription opioid overdose. Opioid-related deaths have increased tenfold since 1999, with no signs of slowing down. The District of Columbia declared a public emergency in November 2023 to draw more attention to the opioid crisis.
In 2023, we are at the precipice of the fourth wave of the crisis, as synthetic opioids like fentanyl are combined with a stimulant, commonly methamphetamine. Speedballs have been common for decades, using stimulants to counterbalance the fatigue that occurs with opiates. The fatal combination of fentanyl and a stimulant was responsible for just 0.6% of overdose deaths in 2010 but 32.3% of opioid deaths in 2021, an over fifty-fold increase in 12 years. Fentanyl, originally used in end-of-life and cancer care, is commonly manufactured in Mexico with precursor chemicals from China. Fentanyl is also commonly added to pressed pills made to look like legitimate prescription medications. In the first nine months of 2023, the Drug Enforcement Agency (DEA) seized over 62 million counterfeit pills and nearly five tons of powdered fentanyl, which equates to over 287 million fatal doses. These staggering seizure numbers do not include local law enforcement efforts, with the New York City Police Department recovering 13 kilos of fentanyl in the Bronx, enough powder to kill 6.5 million people.
The ease of creating and trafficking fentanyl and similar opioids has led to an epidemic in the United States. Currently, fentanyl can be made for pennies and sold for as little as 40 cents in Washington State. The ease of availability has led to deaths in our most vulnerable population—children. Between June and September 2023, there were three fatal overdoses of children five years and younger in Portland, OR. In a high-profile case in New York City, investigators found a kilogram of fentanyl powder in a day care facility after a 1-year-old died and three others became critically ill.
The Biden Administration has responding to the crisis in part by placing sanctions against and indicting executives in Chinese companies for manufacturing and distributing precursor chemicals, which are commonly sold to Mexican drug cartels to create fentanyl. The drug is then trafficked into the United States for sale and use. There are also concerns about fentanyl being used as a weapon of mass destruction, similar to the anthrax concerns in the early 2000s.
The daily concerns of opioid overdoses have plagued public health and law enforcement professionals for years. In Seattle, WA, alone, there are 15 non-fatal overdoses daily, straining the emergency medical systems. There were nearly 5,000 non-fatal overdoses in the first seven months of 2023 in King County, WA, an increase of 70% compared to 2022. In a landmark decision, in March 2023 the Food and Drug Administration (FDA) approved naloxone, a drug to reverse the effects of opioid overdoses, as an over-the-counter nasal spray in an attempt to reduce overdose deaths. Naloxone nasal spray was initially approved for prescription use only in 2015 , significantly limiting access to first responders and available to high-risk patients when prescribed opioids. In New York, physicians have been required to prescribe naloxone to patients at risk of overdose since 2022. Although naloxone is now available without a prescription, access is still limited by price, with one dose costing as much as $65, and some people requiring more than one dose to reverse the overdose. Citing budget concerns, Governor Newsom vetoed California’s proposed AB 1060, which would have limited the cost of naloxone to $10 per dose. Fentanyl testing strips that can be used to test substances for the presence of fentanyl before use show promise in preventing unwanted fentanyl-adulterated overdoses. The Expanding Nationwide Access to Test Strips Act, which was introduced to the Senate in July 2023, would decriminalize the testing strips as an inexpensive way to reduce overdose while following evidence-based harm-reduction theories.
Illicit drugs are also one of the top threats to national security. Law enforcement agencies are dealing with a triple epidemic of gun violence, the opioid crisis, and critical staffing levels. Crime prevention is tied directly to increased police staffing, with lower staffing limiting crime control tactics, such as using interagency task forces, to focus on a specific crime problem. Police are at the forefront of the opioid crisis, expected to provide an emergency response to potential overdoses and ensure public safety while disrupting and investigating drug-related crimes. Phoenix Police Department seized over 500,000 fentanyl pills in June 2023 as part of Operation Summer Shield, showing law enforcement’s central role in fighting the opioid crisis. DHS created a comprehensive interdiction plan to reduce the national and international supply of opioids, working with the private sector to decrease drugs brought into the United States and increasing task forces to focus on drug traffickers.
Prosecutors are starting to charge drug dealers and parents of children exposed to fentanyl in their residences in fatal overdose cases. In an unprecedented action, Attorney General Merrick Garland recently charged Mexican cartel members with trafficking fentanyl and indicting Chinese companies and their executives for creating and selling precursor chemicals. In November 2023, sanctions were placed against the Sinaloa cartel and four firms from Mexico suspected of drug trafficking to the United States, removing their ability to legally access the American banking system. Despite this work, criminal justice-related efforts alone are not reducing overdoses and deaths, showing a need for a multifaceted approach to save lives.
While these numbers of opioid overdoses are appalling, they are likely underreported. Accurate reporting of fatal overdoses varies dramatically across the country, with the lack of training of medicolegal death investigators to recognize potential drug toxicity-related deaths, coupled with the shortage of forensic pathologists and the high costs of toxicology testing, leading to inaccurate cause of death information. The data ecosystem is changing, with agencies and their valuable data remaining disjointed and unable to communicate across systems. A new model could be found in the CDC’s Data Modernization Initiative, which tracked millions of COVID-19 cases across all states and districts, including data from emergency departments and medicolegal offices. This robust initiative to modernize data transfer and accessibility could be transformative for public health. The electronic case reporting system and strong surveillance systems that are now in place can be used for other public health outbreaks, although they have not been institutionalized for the opioid epidemic.
Toxicology testing can take upwards of 8–10 weeks to receive, then weeks more for interpretation and final reporting of the cause of death. The CDC’s State Unintentional Drug Overdose Reporting System receives data from 47 states from death certificates and coroner/medical examiner reports. Even with the CDC’s extensive efforts, the data-sharing is voluntary, and submission is rarely timely enough for tracking real-time outbreaks of overdoses and newly emerging drugs. The increase of novel psychoactive substances, including the addition of the animal tranquilizer xylazineto other drugs, is commonly not included in toxicology panels, leaving early fatal drug interactions undetected and slowing notification of emerging drugs regionally. The data from medicolegal reports is extremely valuable for interdisciplinary overdose fatality review teams at the regional level that bring together healthcare, social services, criminal justice, and medicolegal personnel to review deaths and determine potential intervention points. Overdose fatality review teams can use the data to inform prevention efforts, as has been successful with infant sleeping position recommendations formed through infant mortality review teams.
Plan of Action
Reducing opioid misuse and saving lives requires a multi-stage, multi-agency approach. This includes expanding real-time opioid surveillance efforts; funding for overdose awareness, prevention, and education; and improved training of first responders and medicolegal personnel on recognizing, responding to, and reporting overdoses. Nationwide, improved toxicology testing and reporting is essential for accurate reporting of overdose-involved drugs and determining the efficacy of efforts to combat the opioid epidemic.
Recommendation 1. Fund research to determine the efficacy of current efforts in opioid misuse reduction and prevention.
DOJ should provide grant funding for researchers to outline all known current efforts of opioid misuse reduction and prevention by law enforcement, public health, community programs, and other agencies. The efforts, including the use of suboxone and methadone, should be evaluated to determine if they follow evidence-based practices, how the programs are funded, and their known effect on the community. The findings should be shared widely and without paywalls with practitioners, researchers, and government agencies to hone their future work to known successful efforts and to be used as a foundation for future evidence-based, innovative program implementation.
Recommendation 2. Modernize data systems and surveillance to provide real-time information.
City, county, regional, and state first responder agencies work across different platforms, as do social service agencies, hospitals, private physicians, clinics, and medicolegal offices. A single fatal drug toxicity-related death has associated reports from a law enforcement officer, fire department personnel, emergency medical services, an emergency department, and a medicolegal agency. Additional reports and information are sought from hospitals and clinics, prior treating clinicians, and social service agencies. Even if all of these reports can be obtained, data received and reviewed is not real-time and not accessible across all of the systems.
Medicolegal agencies are arguably the most underprepared for data and surveillance modernization. Only 43% of medicolegal agencies had a computerized case management system in 2018, which was an increase from 31% in 2004. Outside of county or state property, only 75% of medicolegal personnel had internet access from personal devices. The lack of computerized case management systems and limited access to the internet can greatly hinder case reporting and providing timely information to public health and other reporting agencies.
With the availability and use of naloxone by private persons, the Public Naloxone Administration Dashboard from the National EMS Information System (NEMSIS) should be supported and expanded to include community member administration of naloxone. The emergency medical services data can be aligned with the anonymous upload of when, where, and basic demographics for the recipient of naloxone, which can also be made accessible to emergency departments and medicolegal death investigation agencies. While the database likely will not be used for all naloxone administrations, it can provide hot spot information and notify social services of potential areas for intervention and assistance. The database should be tied to the first responder/hospital/medicolegal database to assist in robust surveillance of the opioid epidemic.
Recommendation 3. Increase overdose awareness, prevention education, and availability of naloxone.
Awareness of the likelihood of poisoning and potential death from the use of fentanyl or counterfeit pills is key in prevention. The DEA declared August 21 National Fentanyl Prevention and Awareness Day to increase knowledge of the dangers of fentanyl, with the Senate adopting a resolution to formally recognize the day in 2023. Many states have opioid and fentanyl prevention tactics on their public health websites, and the CDC has educational campaigns designed to reach young adults, though the education needs to be specifically sought out. Funding should be made available to community organizations and city/county governments to create public awareness campaigns about fentanyl and opioid usage, including billboards, television and streaming ads, and highly visible spaces like buses and grocery carts.
ED allows evidence-based prevention programs in school settings to assist in reducing risk factors associated with drug use and misuse. The San Diego Board of Supervisors approved a proposal to add education focused on fentanyl awareness after 12 juveniles died of fentanyl toxicity in 2021. The district attorney supported the education and sought funding to sponsor drug and alcohol training on school campuses. Schools in Arlington, VA, note the rise in overdoses but recognize that preventative education, when present, is insufficient. ED should create prevention programs at grade-appropriate levels that can be adapted for use in classrooms nationwide.
With the legalization of over-the-counter naloxone, funding is needed to provide subsidized or free access to this life-saving medication. Powerful fentanyl analogs require higher doses of naloxone to reverse the toxicity, commonly requiring multiple naloxone administrations, which may not be available to an intervening community member. The State of Washington’s Department of Public Health offers free naloxone kits by mail and at certain pharmacies and community organizations, while Santa Clara University in California has a vending machine that distributes naloxone for free. While naloxone reverses the effects of opioids for a short period, once it wears off, there is a risk of a secondary overdose from the initial ingestion of the opioid, which is why seeking medical attention after an overdose is paramount to survival. Increasing access to naloxone in highly accessible locations—and via mail for more rural locations—can save lives. Naloxone access and basic training on signs of an opioid overdose may increase recognition of opioid misuse and empower the community to provide immediate, lifesaving action.
However, there are concerns that naloxone may end up in a shortage. With its over-the-counter access, naloxone may still be unavailable for those who need it most due to cost (approximately $20 per dose) or access to pharmacies. There is a national push for increasing naloxone distribution, though there are concerns of precursor shortages that will limit or halt production of naloxone. Governmental support of naloxone manufacturing and distribution can assist with meeting demand and ensuring sustainability in the supply chain.
Recommendation 4. Improve training of first responders and medicolegal death investigators.
Most first responders receive training on recognizing signs and symptoms of a potential overdose, and emergency medical and firefighting personnel generally receive additional training for providing medical treatment for those who are under the influence. To avoid exposure to fentanyl, potentially causing a deadly situation for the first responder, additional training is needed about what to do during exposure and how to safely provide naloxone or other medical care. DEA’s safety guide for fentanyl specifically outlines a history of inconsistent and misinformation about fentanyl exposure and treatment. Creating an evidence-based training program that can be distributed virtually and allow first responders to earn continuing education credit can decrease exposure incidents and increase care and responsiveness for those who have overdosed.
While the focus is rightfully placed on first responders as the frontline of the opioid epidemic, medicolegal death investigators also serve a vital function at the intersection of public health and criminal justice. As the professionals who respond to scenes to investigate the circumstances (including cause and manner) surrounding death, medicolegal death investigators must be able to recognize signs of drug toxicity. Training is needed to provide foundational knowledge on deciphering evidence of potential overdose-related deaths, photographing scenes and evidence to share with forensic pathologists, and memorializing the findings to provide an accurate manner of death. Causes of death, as determined by forensic pathologists, need appropriate postmortem examinations and toxicology testing for accuracy, incorporated with standardized wording for death certificates to reflect the drugs contributing to the death. Statistics on drug-related deaths collected by the CDC and public health departments nationwide rely on accurate death certificates to determine trends.
The CDC created the Collaborating Office for Medical Examiners and Coroners (COMEC) in 2022 to provide public health support for medicolegal death investigation professionals. COMEC coordinates health surveillance efforts in the medicolegal community and champions quality investigations and accurate certification of death. The CDC offers free virtual, asynchronous training for investigating and certifying drug toxicity deaths, though the program is not well known or advertised, and there is no ability to ask questions of professionals to aid in understanding the content. Funding is needed to provide no-cost, live instruction, preferably in person, to medicolegal offices, as well as continuing education hours and thorough training on investigating potential drug toxicity-related deaths and accurately certifying death certificates.
Cumulatively, the roughly 2,000 medicolegal death investigation agencies nationwide investigated more than 600,000 deaths in 2018, running on an average budget of $470,000 per agency. Of these agencies, less than 45% had a computerized case management system, which can significantly delay data sharing with public health and allied agencies and reduce reporting accuracy, and only 75% had access to the internet outside of their personally owned devices. Funding is needed to modernize and extend the infrastructure for medicolegal agencies to allow basic functions such as computerized case management systems and internet access, similar to grant funding from the National Network of Public Health Institutes.
Recommendation 5. Fund rapid and thorough toxicology testing in emergency departments and coroner/medical examiner agencies.
Rapid, accurate toxicology testing in an emergency department setting can be the difference between life and death treatment for a patient. Urine toxicology testing is fast, economical, and can be done at the bedside, though it cannot quantify the amount of drug and is not inclusive for emerging drugs. Funding for enhanced accurate toxicology testing in hospitals with emergency departments, including for novel psychoactive substances and opioid analogs, is necessary to provide critical information to attending physicians in a timely manner to allow reversal agents or other vital medical care to be performed.
With the limited resources medicolegal death investigation agencies have nationally and the average cost of $3000 per autopsy performed, administrators need to triage which deaths receive toxicology testing and how in-depth the testing will be. Advanced panels, including ever-changing novel psychoactive substances, are costly and can result in inaccurate cause of death reporting if not performed routinely. Funding should be provided to medicolegal death investigating agencies to subsidize toxicology testing costs to provide the most accurate drugs involved in the death. Accurate cause of death reporting will allow for timely public health surveillance to determine trends and surges of specific drugs. Precise cause of death information and detailed death investigations can significantly contribute to regional multidisciplinary overdose fatality review task forces that can identify potential intervention points to strengthen services and create evidence to build future life-saving action plans.
Recommendation 6. Enhance prevention and enforcement efforts.
DOJ should fund municipal and state law enforcement grants to use evidence-based practices to prevent and enforce drug-related crimes. Grant applications should include a review of the National Institute of Justice’s CrimeSolutions.gov practices in determining potential effectiveness or using foundational knowledge to build innovative, region-specific efforts. The funding should be through competitive grants, requiring an analysis of local trends and efforts and a detailed evaluation and research dissemination plan. Competitive grant funding should also be available for community groups and programs focusing on prevention and access to naloxone.
An often overlooked area of prevention is for justice-involved individuals who enter jail or prison with substance use disorders. Approximately 65% of prisoners in the United States have a substance abuse order, and an additional 20% of prisoners were under the influence of drugs or alcohol when they committed their crime. About 15% of the incarcerated population was formally diagnosed with an opioid use disorder. Medications are available to assist with opioid use disorder treatments that can reduce relapses and post-incarceration toxicity-related deaths, though less than 15% of correctional systems offer medication-assisted opioid use treatments. Extensive case management coupled with trained professionals to prescribe medication-assisted treatment can help reduce opioid-related relapses and overdoses when justice-involved individuals are released to their communities, with the potential to reduce recidivism if treatment is maintained.
DEA should lead local and state law enforcement training on recognizing drug trends, creating regional taskforces for data-sharing and enforcement focus, and organizing drug takeback days. Removing unused prescription medications from homes can reduce overdoses and remove access to unauthorized users, including children and adolescents. Funding to increase collection sites, assist in the expensive process of properly destroying drugs, and advertising takeback days and locations can reduce the amount of available prescription medications that can result in an overdose.
DHS, TREAS, and DOS should expand their current efforts in international trafficking investigations, create additional sanctions against businesses and individuals illegally selling precursor chemicals, and collaborate with countries to universally reduce drug production.
Budget Proposal
A budget of $800 million is proposed to evaluate the current efficacy of drug prevention and enforcement efforts, fund prevention and enforcement efforts, improve training for first responders and medicolegal death investigators, increase rapid and accurate toxicology testing in emergency and medicolegal settings, and enhance collaboration between law enforcement agencies. The foundational research on the efficacy of current enforcement, preventative efforts, and surveillance should receive $25 million, with findings transparently available and shared with practitioners, lawmakers, and community members to hone current practices.
DOJ should receive $375 million to fund grants; collaborative enforcement efforts between local, state, and federal agencies; preventative strategies and programs; training for first responders; and safe drug disposal programs.
CDC should receive $250 million to fund the training of medicolegal death investigators to recognize and appropriately document potential drug toxicity-related deaths, modernize data and reporting systems to assist with accurate surveillance, and provide improved toxicology testing options to emergency departments and medicolegal offices to assist with appropriate diagnoses. Funding should also be used to enhance current data collection efforts with the Overdose to Action program34 by encouraging timely submissions, simplifying the submission process, and helping create or support overdose fatality review teams to determine potential intervention points.
ED should receive $75 million to develop curricula for K-12 and colleges to raise awareness of the dangers of opioids and prevent usage. The curriculum should be made publicly available for access by parents, community groups, and other organizations to increase its usage and reach as many people as possible.
BOP should receive $25 million to provide opioid use disorder medication-assisted treatments by trained clinicians and extensive case management to assist in reducing post-incarceration relapse and drug toxicity-related deaths. The policies, procedures, and steps to create medication-assisted programming should be shared with state corrections departments and county jails to build into their programming to expand use in carceral settings and assist in reducing drug toxicity-related deaths at all incarceration levels.
DOS, DHS, and TREAS should jointly receive $50 million to strengthen their current international investigations and collaborations to stop drug trafficking, the manufacture and sales of precursors, and combating organized crime’s association with the illegal drug markets.
Conclusion
Opioid-related overdoses and deaths continue to needlessly and negatively affect society, with parents burying children, sometimes infants, in an unnatural order. With the low cost of fentanyl production and the high return on investment, fentanyl is commonly added to illicit drugs and counterfeit, real-looking prescription pills. Opioid addiction and fatal overdoses affect all genders, races, ethnicities, and socioeconomic statuses, with no end to this deadly path in sight. Combining public health surveillance with enforcement actions, preventative education, and innovative programming is the most promising framework for saving lives nationally.
Opioid overdoses are occurring all over the nation, including rural areas and tribal communities. Some states have dashboards showing opioid-related deaths by county, similar to the Missouri Department of Health and Senior Services, as do some local county-level health departments like the Washtenaw County, MI Health Department. Mapping programs, such as ODMAP, are available to public safety and public health agencies to watch near-real-time overdose reports, and community organizations may also be tracking overdoses with publicly available information. The CDC’s Overdose Data to Action Program provides data from 47 states and the District of Columbia, producing a robust dashboard separated by participating states and including information about circumstances surrounding deaths and opportunities for intervention.
Community groups can work to spread awareness of opioid dangers and provide preventative education. The DEA has social media resources and a partner toolbox to increase awareness about counterfeit prescription drugs. The National Harm Reduction Coalition has fact sheets and a resource library with webinars and training guides to assist with awareness and prevention campaigns. Community members can also advocate for awareness and preventive education to be added to local K-12 and college curricula. Other key actions are outreach to at-risk populations and empowering parents and guardians to discuss the dangers of opioids with their children.
In 2019, there were approximately 600,000 deaths worldwide related to drug toxicity, with about 80% involving opioids. The United States had 70,630 drug toxicity-related deaths in 2019, 70.6% of which involved opioids, making the country responsible for about 12% of drug-related deaths worldwide. Overdose rates in the United States are significantly overrepresented in drug-related deaths compared to the international population, though data collection and reporting in other countries may not be as robust.
Prior funding to address the opioid epidemic has shown researchers and practitioners what has and has not worked. Despite extensive funding, enacting the National Guard, and creating task forces to combat fentanyl opioid-related overdoses, San Francisco reported 692 drug toxicity-related deaths from January to October 2023, surpassing the 649 deaths in 2022 and the 642 deaths in 2021. San Francisco is on track to have nearly 70 deaths per month, with the final total likely increasing to over 800 by the end of 2023. While this is only one example, the CDC shows an upward predicted value of drug toxicity-related deaths throughout 2023 using national data.
The current funding requests and structure will help to bring forward the dark figures of the epidemic and build robust surveillance systems to track opioid-related toxicities in real time. There are tools available from the pandemic and past opioid use reduction efforts that can be tailored to data collection for opioid-related morbidity and mortality, which, combined with other strategies, can end the opioid epidemic. The increase in overdose awareness and education may be the key to a reduction in overdoses and deaths, similar to how education assisted in curbing human immunodeficiency virus (HIV) transmission. Viewing the epidemic through a public health lens and coupling a pulling-levers approach to crime prevention with educational and data components has the potential to save a significant number of lives.
Tracking Extreme Heat Federal Policy and Funding
Last year was the hottest year in recorded human history. In summer 2023 alone, up to 275 million Americans were placed under some type of heat advisory. Experts at NOAA project a one-in-three chance that 2024 will be even warmer than 2023 — with a 99% chance that 2024 will rank among the top five warmest years. With “danger season” 2024, the time when extreme heat and numerous other climate-related hazards in the United States tend to occur — beginning after April 29th, there is a vital need to build resilience to the impending heat waves.
To begin to respond to this urgent need at the federal level, FAS engaged +85 federal policy experts and recruited 33 authors to work on +18 policy memos through our Extreme Heat Policy Sprint, generating +150 policy recommendations to address extreme heat’s impacts and build community resilience. Our contributors’ recommendations represent the building blocks of a whole-of-government strategy on extreme heat, spanning six domains:
- Infrastructure and the built environment
- Workforce safety and development
- Public health, medical preparedness, and health security
- Food security and multi-hazard resilience
- Planning and response
- Data and indices
Collectively, FAS has identified 34 offices and/or agencies that can act on extreme heat. However, as noted in our previous publication, extreme heat receives minimal targeted federal support and funding for planning, mitigation, and recovery despite being the number one weather-related killer of Americans. The national response to extreme heat is still being developed and requires increased, coordinated action across the White House, Congress, and federal agencies. Improved coordination and effective planning requires a clear understanding of the landscape of the existing federal efforts. For this reason, the Federation of American Scientists has put together an Extreme Heat Federal Policy and Funding tracker to monitor the progress of federal actions on extreme heat, enhance accountability, and to allow stakeholders to stay informed on the evolving state of U.S. climate-change resilience response as it evolves. This tracker is organized around our six key domains of federal opportunity.
In the absence of a national strategy, states, counties, and cities around the country have had to take on the responsibility of experimenting and attempting to address extreme heat in their communities with limited available resources. While many state and local governments are working diligently to make significant advances, national extreme heat resilience requires a whole-of-government federal approach, as it directly impacts public health, energy, housing, national security, international relations, and many more policy domains. The federal government plays a critical role in scaling heat resilience interventions through funding, guidance, research and development, regulations, and other policy levers.
Executive branch agencies need a government-wide coordination strategy to prioritize and address extreme heat nationwide. This strategy requires comprehensive reviews of available resources for financial assistance, assessments of regulatory and rulemaking authority, and an emphasis on legislative action — in order to define the problems to solve, assign priorities for agencies, and develop evaluation metrics for review, adjustment, and renewal of programs The FAS Federal Extreme Heat Policy and Funding tracker serves as a key starting point towards these necessary actions.
Defining Disaster: Incorporating Heat Waves and Smoke Waves into Disaster Policy
Extreme heat – and similar people-centered disasters like heavy wildfire smoke – kills thousands of Americans annually, more than any other weather disaster. However, U.S. disaster policy is more equipped for events that damage infrastructure than those that mainly cause deaths. Policy actions can save lives and money by better integrating people-centered disasters.
Challenge and Opportunity
At the federal level, emergency management is coordinated through the Federal Emergency Management Agency (FEMA), with many other agencies as partners, including Centers for Disease Control (CDC), Department of Housing and Urban Development (HUD), and Small Business Administration (SBA). Central to the FEMA process is the requirement under the Stafford Act that the President declare a major disaster, which has never happened for extreme heat. This seems to be caused by a lack of tools to determine when a heat wave event escalates into a heat wave disaster, as well as a lack of a clear vision of federal responsibilities around a heat wave disaster.
Gap 1. When is a heat event a heat disaster?
A core tenet of emergency management is that events escalate into disasters when the impacts exceed available resources. Impact measurement is increasingly quantitative across FEMA programs, including quantitative metrics used in awarding Fire Management Assistance Grant (FMAG), Public Assistance (PA), and Individual Assistance (IA) and in the Benefit Cost Analysis (BCA) for hazard mitigation grants.
However, existing calculations are unable to incorporate the health impacts that are a main impact of heat waves. When health impacts are included in a calculation, it is only in limited cases; for example, the BCA allows mental healthcare savings, but only for residential mitigation projects that reduce post-disaster displacement.
Gap 2. What is the federal government’s role in a heat disaster?
Separate from the declaration of a major disaster is the federal government’s role during that disaster. Existing programs within FEMA and its partner agencies are designed for historic disasters rather than those of the modern and future eras. For example, the National Risk Index (NRI), used to understand the national distribution of risks and vulnerability, bases its risk assessment on events between 1996 and 2019. As part of considering future disasters, disaster policy should consider intensified extreme events and compound hazards (e.g., wildfire during a heat wave) that are more likely in the future.
A key part of including extreme heat and other people-centered disasters will be to shift toward future-oriented resilience and adaptation. FEMA has already been making this shift, including a reorganization to highlight resilience. The below plan of action will further help FEMA with its mission to help people before, during, and after disasters.
Plan of Action
To address these gaps and better incorporate extreme heat and people-centered disasters into U.S. emergency management, Congress and federal agencies should take several interrelated actions.
Recommendation 1. Defining disaster
To clarify that extreme heat and other people-centered disasters can be disasters, Congress should:
(1) Add heat, wildfire smoke, and compound events (e.g., wildfire during a heat wave) to the list of disasters in Section 102(2) of the Stafford Act. Though the list is intended to be illustrative rather than exhaustive, as demonstrated by the declaration of COVID-19 as a disaster despite not being on the list, explicit inclusion of these other disasters on the list clarifies that intent. This action is widely supported and example legislation includes the Extreme Heat Emergency Act of 2023.
(2) FEMA should standardize procedures for determining when disparate events are actually a single compound event. For example, many individual tornadoes in Kentucky in 2021 were determined to be the results of a single weather pattern, so the event was declared as a disaster, but wildfires that started due to a single heat dome in 2022 were determined to be individual events and therefore unable to receive a disaster declaration. Compound hazards are expected to be more common in the future, so it is critical to work toward standardized definitions.
(3) Add a new definition of “damage” to Section 102 of the Stafford Act that includes human impacts such as death, illness, economic impacts, and loss of critical function (i.e., delivery of healthcare, school operations, etc.). Including this definition in the statute facilitates the inclusion of these categories of impact.
To quantify the impacts of heat waves, thereby facilitating disaster decisions, FEMA should adopt strategies already used by the federal government. In particular, FEMA should:
(4) Work with HHS to expand the capabilities of the National Syndromic Surveillance Program (NSSP) to evaluate in real time various societal impacts like the medical-care usage and work or school days lost. Recent studies indicate that lost work productivity is a major impact of extreme heat that is currently unaccounted—a gap of potentially billions of dollars. The NSSP Community of Practice can help expand tools across multiple jurisdictions too. Expanding syndromic surveillance expands our ability to measure the impacts of heat, building on the tools available through the CDC Heat and Health Tracker.
(5) Work with CDC to expand their use of excess-death and flu-burden methods, which can provide official estimates of the health impacts of extreme heat. These methods are already in use for heat, but should be regularly applied at the federal level, and would complement the data available from health records via NSSP because it calculates missing data.
(6) Work with EPA to expand BenMAP software to include official estimates of health impacts attributable to extreme heat. The current software provides official estimates of health impacts attributable to air pollution and is used widely in policy. Research is needed to develop health-impact functions for extreme heat, which could be solicited in a research call such as through NIH’s Climate and Health initiative, conducted by CDC epidemiologists, added to the Learning Agenda for FEMA or a partner agency, or tasked to a national lab. Additional software development is also needed to cover real-time and forecast impacts in addition to the historic impacts it currently covers. The proposed tool complements Recommendations #4-5 because it includes forecast data.
(7) Quantify heat illness and death impacts. Real-time data is available in the CDC Heat and Health Tracker. These impacts can be converted to dollars for comparison to property damage using the Value of a Statistical Life (VSL), which FEMA already does in the NRI ($11.6 million per death and $1.16 million per injury in 2022). VSL should be expanded across FEMA programs, in particular the decision for major disaster declarations. VSL could be immediately applied to current data from NSSP, to expanded NSSP and excess-death data (Recommendations #4-5), and is already incorporated into BenMAP so would be available in the expanded BenMAP (Recommendation #6).
(8) Quantify the impact of extreme heat on critical infrastructure, including agriculture. Improved quantification methods could be achieved by expanding the valuation methods for infrastructure damage already in the NRI and could be integrated with the National Integrated Heat Health Information System (NIHHIS). The damage and degradation of infrastructure is often underestimated and should be accurately quantified. For example,
- (a) The Federal Highway Administration (FHWA) has reported on extreme heat and documented heat impacts on asset management, where the Department of Transportation (DOT) in multiple states, including Arizona’s DOT, and the National Cooperative Highway Research Program have developed evaluation methods for quantifying impacts, and
- (b) Impacts to critical infrastructure have been quantified across sectors and should include representative agencies such as transportation (e.g., FHWA, DOT), transit (e.g., Federal Transit Authority (FTA)), energy (e.g., Department of Energy (DOE), EPA, water/sanitation (e.g., U.S. Army Corps of Engineers (USACE), Bureau of Reclamation (BOR)), telecommunications (e.g., Department of Homeland Security, Federal Communications Commission), social (e.g., HUD, HHS, Department of Education (ED)), and environmental infrastructures (e.g., EPA, USACE, National Parks Service (NPS)).
Together, these proposed data tools would provide FEMA with a comprehensive understanding of the impacts of extreme heat on human health in the past, present, and near future, putting heat on the same playing field as other disasters.
Real-time impacts are particularly important for FEMA to investigate requests for a major disaster declaration. Forecast impacts are important for the ability to preposition resources, as currently done for hurricanes. The goal for forecasting should be 72 hours. To achieve this goal from current models (e.g., air quality forecasts are generally just one day in advance):
(9) Congress should fund additional sensors for extreme weather disasters, to be installed by the appropriate agencies. More detailed ideas can be found in other FAS memos for extreme heat and wildfire smoke and in recommendation 44 of the recent Wildland Fire Commission report.
(10) Congress should invest in research on integrated wildfire-meteorological models through research centers of excellence funded by national agencies or national labs. Federal agencies can also post specific questions as part of their learning agendas. Models should specifically record the contribution of wildfire smoke from each landscape parcel to overall air pollution in order to document the contribution of impacts. This recommendation aligns with the Fire Environment Center proposed in the Wildland Fire Commission report.
Recommendation 2. Determining federal response to heat disasters
To incorporate extreme heat and people-centered disasters across emergency management, FEMA and its peer agencies can expand existing programs into new versions that incorporate such disasters. We split these programs here by the phase of emergency management.
Preparedness
(11) Using Flood Maps as a model, FEMA should create maps for extreme heat and other people-centered disasters. Like flood maps, these new maps should highlight the infrastructure at risk of failure or the loss of access to critical infrastructure (e.g., FEMA Community Lifelines) during a disaster. Failure here is defined as the inability of infrastructure to provide its critical function(s); infrastructure that ceases to be usable for its purpose when an extreme weather event occurs (i.e., bitumen softening on airport tarmacs, train line buckling, or schools canceled because classrooms were too hot or too smokey). This includes impacts to evacuation routes and critical infrastructure that would severely impact the functioning of society. Creating such a map requires a major interagency effort integrating detailed information on buildings, heat forecasts, energy grid capacity, and local heat island maps, which likely requires major interagency collaboration. NIHHIS has most of the interagency collaborators needed for such effort, but should also include the Department of Education. Such an effort likely will need direct funding from Congress in order to have the level of effort needed.
(12) FEMA and its partners should publish catastrophic location-specific scenarios to align preparedness planning. Examples include the ARkStorm for atmospheric rivers, HayWired for earthquake, and Cascadia Rising for tsunami. Such scenarios are useful because they help raise public awareness and increase and align practitioner preparedness. A key part of a heat scenario should be infrastructure failure and its cascading impacts; for example, grid failure and the resulting impact on healthcare is expected to have devastating effects.
(13) FEMA should incorporate future projections of disasters into the NRI. The NRI currently only uses historic data on losses (typically 1996 to 2019). An example framework is the $100 million Prepare California program, which combined historic and projected risks in allocating preparedness funds. An example of the type of data needed for extreme heat includes the changes in extreme events that are part of the New York State Climate Impacts Assessment.
(14) FEMA should expand its Community Lifelines to incorporate extreme heat and cascading impacts for critical infrastructure as a result of extreme heat, which must remain operable during and after a disaster to avoid significant loss of human life and property.
(15) The strategic national stockpile (SNS) should be expanded to focus on tools that are most useful in extreme weather disasters. A key consideration will be fluids, including intravenous (IV) fluids, which the current medical-focused SNS excludes due to weight. In fact, the SNS relies on the presence of IV fluids at the impacted location, so if there is a shortage due to extreme heat, additional medicines might not be deliverable. To include fluids, a new model will be necessary because of the logistics of great weight.
(16) OSHA should develop occupational safety guidelines to protect workers and students from hazardous exposures, expanding on its outdoor and indoor heat-related hazards directive. Establishing these thresholds, such as max indoor air temperatures similar to California’s Occupational Safety and Health Standards Board, can help define the threshold of when a weather event escalates into a disaster. No federal regulations exist for air quality, so California’s example could be used as a template. The need already exists: an average of 2,700 heat-related injuries and 38 heat-related fatalities were reported annually to OSHA between 2011 and 2019.
(17) FEMA and its partners should expand support for community-led multi-hazard resilience hubs, including learning from those focused on extreme heat. FEMA already has its Hubs for Equitable Resilience and Engagement, and EPA has major funding available to support resilience hubs. This equitable model of disaster resilience that centers on the needs of the specific community should be supported.
Response
(18) FEMA should introduce smaller disaster-assistance grants for extreme weather disasters: HMAG, CMAG, and SMAG (Heat, Cold, and Smoke Management Assistance Grants, respectively). They should be modeled on FMAG grants, which are rapidly awarded when firefighting costs exceed available resources but do not necessarily escalate to the level of a major disaster declaration. For extreme weather disasters, the model would be similar, but the eligible activities might focus on climate-controlled shelters, outreach teams to reach especially vulnerable populations, or a surge in medical personnel and equipment. Just like firefighting equipment and staff needed to fight wildfires, this equipment and staff are needed to reduce the impacts of the disaster. FMAG is supported by the Disaster Relief Fund, so if the H/C/SMAG programs also tap that, it will require additional appropriations. Shelters are already supported by the Public Assistance (PA) program, but PA requires a major disaster declaration, so the introduction of lower-threshold funds would increase access.
(19) HHS could activate Disaster Medical Assistance Teams to mitigate any surge in medical needs. These teams are intended to provide a surge in medical support during a disaster and are deployed in other disasters. See our other memos on this topic.
(20) FEMA could deploy Incident Management Assistance Teams and supporting gear for additional logistics. They can also deploy backup energy resources such as generators to prevent energy failure at critical infrastructure.
Recovery and Mitigation
(21) Programs addressing gray or green infrastructure should consider the impact upgrades will have on heat mitigation. For example, EPA and DOE programs funding upgrades to school gray infrastructure should explicitly consider whether proposed upgrades will meet the heat mitigation needs based on climate projections. Projects funding schoolyard redesign should explicitly consider heat when planning blacktop, playground, and greenspace placement to avoid accidentally creating hot spots where children play. CAL FIRE’s grant to provide $47 million for schools to convert asphalt to green space is a state-level example.
(22) Expand the eligible actions of FEMA’s Hazard Mitigation Assistance (HMA) to include installation/upgrade of heating, ventilation, and cooling (HVAC) systems and a more expansive program to support nature-based solutions (NBS) like green space installation. Existing guidance allows HVAC mitigation for other hazards and incentivizes NBS for other hazards.
(23) Increase alignment across federal programs, identifying programs where goals align. For example, FEMA just announced that solar panels would be eligible for the 75% federal cost share as part of mitigation programs; other climate and weatherization improvements should also be eligible under HMA funds.
(24) FEMA should modify its Benefit Cost Analysis (BCA) process to fairly evaluate mitigation of health and life-safety hazards, to better account for mitigation of multiple hazards, and to address equity considerations introduced in Office of Management and Budget’s recent BCA proposal. Some research is likely needed (e.g., the cost-effectiveness of various nature-based solutions like green space is not yet well-defined enough to use in a BCA); this research could be performed by national labs, put into FEMA’s Learning Agenda, or tasked to a partner agency like DOE.
(25) Expand the definition of medical devices to include items that protect against extreme weather. For example, the Center for Medicare and Medicaid Services could define air-conditioning units and innovative personal cooling devices as eligible for prescription under Medicare/Medicaid.
To support the above recommendations, Congress should:
(26) Ensure FEMA is sufficiently and consistently funded to conduct resilience and adaptation activities. Congress augments the Disaster Relief Fund in response to disasters, but they report that the fund will be billions of dollars in deficit by September 2024. It has furthermore been reported that FEMA has delayed payments due to uncertainty of funding through Congressional budget negotiations. In order to support the above programs, it is essential that Congress fund FEMA at a level needed to act. To support FEMA’s shift to a focus on resilience, the increase in funding should be through annual appropriations rather than the Disaster Relief Fund, which is augmented on an ad hoc basis.
(27) Convene a congressional commission like the recent Wildland Fire Commission to analyze the federal capabilities around extreme weather disasters and/or extreme heat. This commission would help source additional ideas and identify political pathways toward creating these solutions, and is merited by the magnitude of the disaster.
Conclusion
People across the U.S. are being increasingly exposed to extreme heat and other people-centered disasters. The suggested policies and programs are needed to upgrade national emergency management for the modern and future era, thereby saving lives and reducing disaster costs to the public.
We estimate a minimum of 1,670 deaths and $157.8 billion of annual heat impacts. These deaths and dollar amounts exceed almost every recorded disaster in U.S. history. Only COVID-19, 9/11, and Hurricanes Maria and Katrina have more deaths, and only Hurricanes Katrina and Harvey have caused more dollar damage. It should be noted that most of the estimates reported are several years out of date and exclude major heat waves of 2021 and 2022. For example, individual heat waves produced sizable numbers of deaths, including 395 deaths in a 2022 California heat wave and 600 deaths in the 2021 Pacific Northwest heatwave.
- Health ($20 billion): The CDC’s 2022 Mortality Statistics estimates 1,670 deaths annually are attributable to extreme heat. Using FEMA’s estimate of the Value of a Statistical Life ($11.6 million 2022 USD), that is $19.4 billion. Additional costs come from the burden on the healthcare system, which is estimated at $1 billion annually.
- Infrastructure ($1–16 billion): The impacts of extreme heat are estimated at $1 billion in 2020 and are projected to impose a cost burden of approximately $26 billion on the U.S. transportation sector by the year 2040. Agriculture loses billions of dollars each year from natural hazards. Drought, made worse by high heat conditions, accounts for a significant amount of the losses. In 2023, 80% of emergency disaster designations declared by the United States Department of Agriculture (USDA) were for drought or excessive heat. In 2023, the Southern/Midwestern drought and heatwave was the costliest disaster, at $14.5 billion in losses.
- Workforce ($136.8 billion annually): Between 2011 and 2019, an average of 2,700 heat-related injuries and 38 heat-related fatalities were reported annually to OSHA ($36.8 billion annually using the VSL). Lost work productivity is a major impact of extreme heat that is currently unaccounted for, but was estimated at $100 billion for 2020 and is projected to reach $500 billion by 2050.
- Other people-centered disasters: There are an estimated 6,000 annual acute and chronic deaths due to wildfire smoke (spanning 2006–2018) and 1,300 deaths attributable to extreme cold (estimates prior to 2010). These health impacts are comparable to those of extreme heat, and again even these high impacts exclude recent smoke waves of 2020, 2021, and 2023 and cold waves of 2021 and 2022.
It is insufficient to just add heat to the list of disasters enumerated in the Stafford Act because it omits (1) the important recognition of compound events that often are associated with extreme heat, (2) other people-centered disasters like smoke waves, and (3) the ability to measure these disasters. We, therefore, recommend some version of the following text:
Section 102(2) of the Robert T. Stafford Disaster Relief and Emergency Assistance Act (42 U.S.C. 5122(2)) is amended by striking “or drought” and inserting “drought, heat, smoke, or any other weather pattern causing a combination of the above”.
Section 102 of the Robert T. Stafford Disaster Relief and Emergency Assistance Act (42 U.S.C. 5122(2)) is amended by inserting
(13) DAMAGE—“Damage” means–
- (A) Loss of life or health impacts requiring medical care
- (B) Loss of property or impacts on property reducing its ability to function
- (C) Diminished usable lifespan for infrastructure
- (D) Economic damage, which includes the value of a statistical life, burden on the healthcare system due to injury, burden on the economy placed by lost days of work or school, agricultural losses, or any other economic damage that is directly measurable or calculated.
- (E) Infrastructure failure of any duration, including temporary, that could lead to any of the above
Enhancing Public Health Preparedness for Climate Change-Related Health Impacts
The escalating frequency and intensity of extreme heat events, exacerbated by climate change, pose a significant and growing threat to public health. This problem is further compounded by the lack of standardized education and preparedness measures within the healthcare system, creating a critical gap in addressing the health impacts of extreme heat. The Department of Health and Human Services (HHS), especially the Centers for Medicare & Medicaid Services (CMS), the Health Resources and Services Administration (HRSA), and the Office of Climate Change and Health Equity (OCCHE) can enhance public health preparedness for the health impacts of climate change. By leveraging funding mechanisms, incentives, and requirements, HHS can strengthen health system preparedness, improve health provider knowledge, and optimize emergency response capabilities.
By focusing on interagency collaboration and medical education enhancement, strategic measures within HHS, the healthcare system can strengthen its resilience against the health impacts of extreme heat events. This will not only improve coding accuracy, but also enhance healthcare provider knowledge, streamline emergency response efforts, and ultimately mitigate the health disparities arising from climate change-induced extreme heat events. Key recommendations include: establishing dedicated grant programs and incentivizing climate-competent healthcare providers; integrating climate-resilience metrics into quality measurement programs; leveraging the Health Information Technology for Economic and Clinical Health (HITECH) Act to enhance ICD-10 coding education; and collaborating with other federal agencies such as the Department of Veterans Affairs (VA), the Federal Emergency Management Agency (FEMA), and the Department of Defense (DoD) to ensure a coordinated response. The implementation of these recommendations will not only address the evolving health impacts of climate change but also promote a more resilient and prepared healthcare system for the future.
Challenge
The escalating frequency and intensity of extreme heat events, exacerbated by climate change, pose a significant and growing threat to public health. The scientific consensus, as documented by reports from the Intergovernmental Panel on Climate Change (IPCC) and the National Climate Assessment, reveals that vulnerable populations, such as children, pregnant people, the elderly, and marginalized communities including people of color and Indigenous populations, experience disproportionately higher rates of heat-related illnesses and mortality. The Lancet Countdown’s 2023 U.S. Brief underscores the escalating threat of fossil fuel pollution and climate change to health, highlighting an 88% increase in heat-related mortality among older adults and calling for urgent, equitable climate action to mitigate this public health crisis.
Inadequacies in Current Healthcare System Response
Reports from healthcare institutions and public health agencies highlight how current coding practices contribute to the under-recognition of heat-related health impacts in vulnerable populations, exacerbating existing health disparities. The current inadequacies in ICD-10 coding for extreme heat-related health cases hinder effective healthcare delivery, compromise data accuracy, and impede the development of targeted response strategies. Challenges in coding accuracy are evident in existing studies and reports, emphasizing the difficulties healthcare providers face in accurately documenting extreme heat-related health cases. An analysis of emergency room visits during heat waves further indicates a gap in recognition and coding, pointing to the need for improved medical education and coding practices. Audits of healthcare coding practices reveal inconsistencies and inaccuracies that stem from a lack of standardized medical education and preparedness measures, ultimately leading to underreporting and misclassification of extreme heat cases. Comparative analyses of health data from regions with robust coding practices and those without highlight the disparities in data accuracy, emphasizing the urgent need for standardized coding protocols.
There is a crucial opportunity to enhance public health preparedness by addressing the challenges associated with accurate ICD-10 coding in extreme heat-related health cases. Reports from government agencies and economic research institutions underscore the economic toll of extreme heat events on healthcare systems, including increased healthcare costs, emergency room visits, and lost productivity due to heat-related illnesses. Data from social vulnerability indices and community-level assessments emphasize the disproportionate impact of extreme heat on socially vulnerable populations, highlighting the urgent need for targeted policies to address health disparities.
Opportunity
As Medicare is the largest federal source of Graduate Medical Education (GME) funding (Figure 1), the Department of Health and Human Services’ (HHS) Centers for Medicare & Medicaid Services (CMS) and the National Center for Health Statistics (NCHS) play a critical role in developing coding guidelines. Thus, it is essential for HHS, CMS, and other pertinent coordinating agencies to be involved in the process for developing climate change-informed graduate medical curricula.
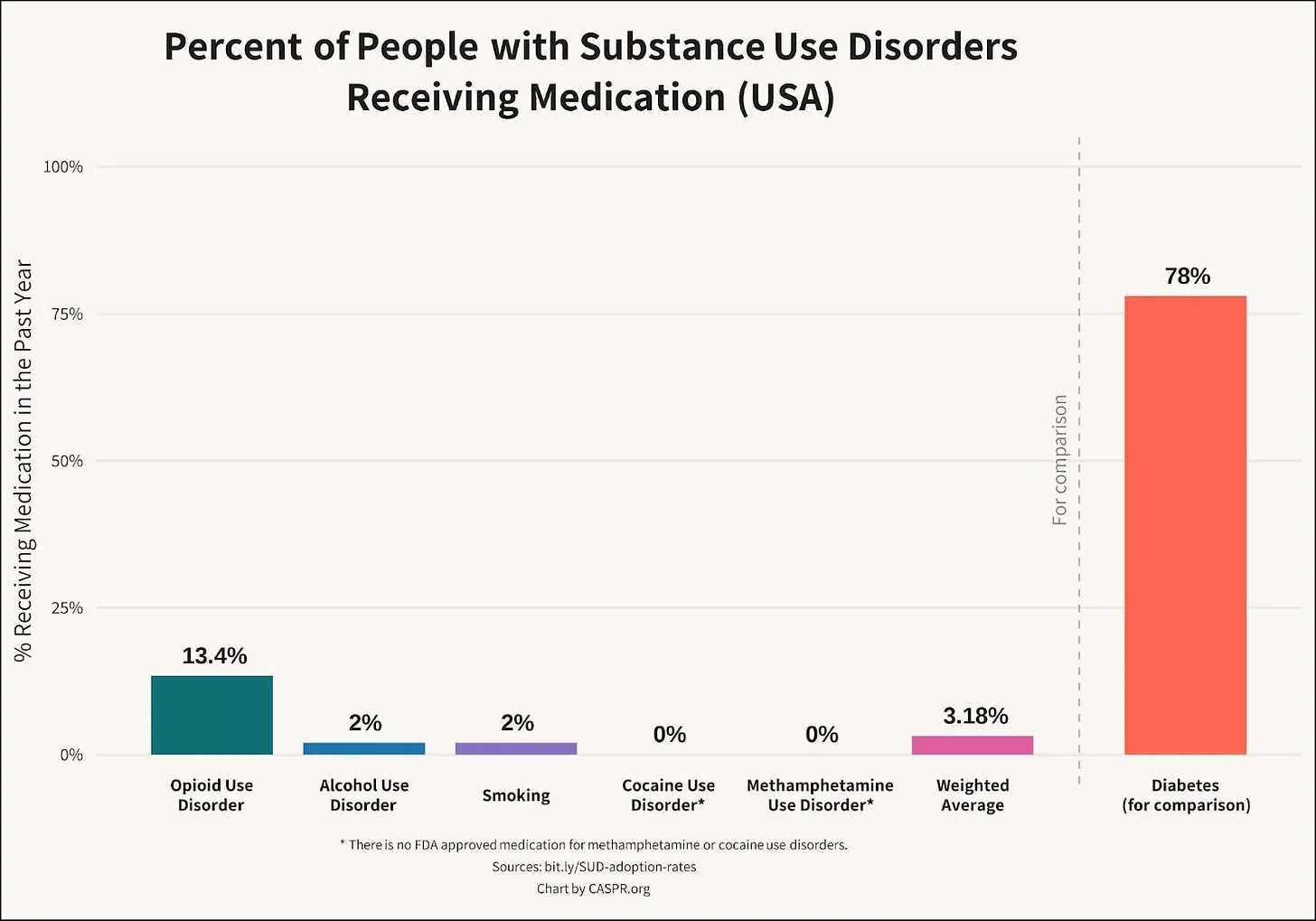{kind=link}
By focusing on medical education enhancement, strategic measures within HHS, and fostering interagency collaboration, the healthcare system can strengthen its resilience against the health impacts of extreme heat events. Improving coding accuracy, enhancing healthcare provider knowledge, streamlining emergency response efforts, and mitigating health disparities related to extreme heat events will ultimately strengthen the healthcare system and foster more effective, inclusive, and equitable climate and health policies. Improving the knowledge and training of healthcare providers empowers them to respond more effectively to extreme heat-related health cases. This immediate response capability contributes to the overarching goal of reducing morbidity and mortality rates associated with extreme heat events and creates a public health system that is more resilient and prepared for emerging challenges.
The inclusion of ICD-10 coding education into graduate medical education funded by CMS aligns with the precedent set by the Pandemic and All Hazards Preparedness Act (PAHPA), emphasizing the importance of preparedness and response to public health emergencies. Similarly, drawing inspiration from the Health Information Technology for Economic and Clinical Health Act (HITECH Act), which promotes the adoption of electronic health records (EHR) systems, presents an opportunity to modernize medical education and ensure the seamless integration of climate-related health considerations. This collaborative and forward-thinking approach recognizes the interconnectedness of health and climate, offering a model that can be applied to various health challenges. Integrating mandates from PAHPA and the HITECH Act serves as a policy precedent, guiding the healthcare system toward a more adaptive and proactive stance in addressing climate change impacts on health.
Conversely, the consequences of inaction on the health impacts of extreme heat extend beyond immediate health concerns. They permeate through the fabric of society, widening health disparities, compromising the accuracy of health data, and undermining emergency response preparedness. Addressing these challenges requires a proactive and comprehensive approach to ensure the well-being of communities, especially those most vulnerable to the effects of extreme heat.
Plan of Action
The following recommendations aim to facilitate public health preparedness for extreme heat events through enhancements in medical education, strategic measures within the Department of Health and Human Services (HHS), and fostering interagency collaboration.
Recommendation 1a. Integrate extreme heat training into the GME curriculum.
Integrating modules on extreme heat-related health impacts and accurate ICD-10 coding into medical education curricula is essential for preparing future healthcare professionals to address the challenges posed by climate change. This initiative will ensure that medical students receive comprehensive training on identifying, treating, and documenting extreme heat-related health cases. Sec. 304. Core Education and Training of the PAHPA provides policy precedent to develop foundational health and medical response curricula and training materials by modifying relevant existing programs to enhance responses to public health emergencies. Given the prominence of Medicare in funding medical residency training, policies that alter Medicare GME can affect the future physician supply and can be used to address identified healthcare workforce priorities related to extreme heat (Figure 2).
Figure 2: A model for comprehensive climate and medical education (adapted from Jowell et al. 2023)
Recommendation 1b. Collaborate with Veterans Health Administration Training Programs.
Partnering with the Department of Veterans Affairs (VA) to extend climate-related health coding education to Veterans Health Administration (VHA) training programs will enhance the preparedness of healthcare professionals within the VHA system to manage and document extreme heat-related health cases among veteran populations.
Recommendation 2. Collaborate with the Agency for Healthcare Research and Quality (AHRQ)
Establishing a collaborative research initiative with the Agency for Healthcare Research and Quality (AHRQ) will facilitate the in-depth exploration of accurate ICD-10 coding for extreme heat-related health cases. This should be accomplished through the following measures:
Establish joint task forces. CMS, NCHS, and AHRQ should establish joint research initiatives focused on improving ICD-10 coding accuracy for extreme heat-related health cases. This collaboration will involve identifying key research areas, allocating resources, and coordinating research activities. Personnel from each agency, including subject matter experts and researchers from the EPA, NOAA, and FEMA, will work together to conduct studies, analyze data, and publish findings. By conducting systematic reviews, developing standardized coding algorithms, and disseminating findings through AHRQ’s established communication channels, this initiative will improve coding practices and enhance healthcare system preparedness for extreme heat events.
Develop standardized coding algorithms. AHRQ, in collaboration with CMS and NCHS, will lead efforts to develop standardized coding algorithms for extreme heat-related health outcomes. This involves reviewing existing coding practices, identifying gaps and inconsistencies, and developing standardized algorithms to ensure consistent and accurate coding across healthcare settings. AHRQ researchers and coding experts will work closely with personnel from CMS and NCHS to draft, validate, and disseminate these algorithms.
Integrate into Continuous Quality Improvement (CQI) programs. Establish collaborative partnerships between the VA and other federal healthcare agencies, including CMS, HRSA, and DoD, to integrate education on ICD-10 coding for extreme heat-related health outcomes into CQI programs. Regularly assess the effectiveness of training initiatives and adjust based on feedback from healthcare providers. For example, CMS currently requires physicians to screen for the social determinants of health and could include level of climate and/or heat risk within that screening assessment.
Allocate resources. Each agency will allocate financial resources, staff time, and technical expertise to support collaborative activities. Budget allocations will be based on the scope and scale of specific initiatives, with funds earmarked for research, training, data sharing, and evaluation efforts. Additionally, research funding provided through PHSA Titles VII and VIII can support studies evaluating the effectiveness of educational interventions on climate-related health knowledge and practice behaviors among healthcare providers.
Recommendation 3. Leverage the HITECH Act and EHR.
Recommendation 4. Establish climate-resilient health system grants to incentivize state-level climate preparedness initiatives
HHS and OCCHE should create competitive grants for states that demonstrate proactive climate change adaptation efforts in healthcare. These agencies can encourage states to integrate climate considerations into their health plans by providing additional funding to states that prioritize climate resilience.
Within CMS, the Center for Medicare and Medicaid Innovation (CMMI) could help create and administer these grants related to climate preparedness initiatives. Given its focus on innovation and testing new approaches, CMMI could design grant programs aimed at incentivizing state-level climate resilience efforts in healthcare. Given its focus on addressing health disparities and promoting preventive care, the Bureau of Primary Health Care (BPHC) within HRSA could oversee grants aimed at integrating climate considerations into primary care settings and enhancing resilience among vulnerable populations.
Conclusion
These recommendations provide a comprehensive framework for HHS — particularly CMS, HRSA, and OCCHE— to bolster public health preparedness for the health impacts of extreme heat events. By leveraging funding mechanisms, incentives, and requirements, HHS can enhance health system preparedness, improve health provider knowledge, and optimize emergency response capabilities. These strategic measures encompass a range of actions, including establishing dedicated grant programs, incentivizing climate-competent healthcare providers, integrating climate-resilience metrics into quality measurement programs, and leveraging the HITECH Act to enhance ICD-10 coding education. Collaboration with other federal agencies further strengthens the coordinated response to the growing challenges posed by climate change-induced extreme heat events. By implementing these policy recommendations, HHS can effectively address the evolving landscape of climate change impacts on health and promote a more resilient and prepared healthcare system for the future.
This idea of merit originated from our Extreme Heat Ideas Challenge. Scientific and technical experts across disciplines worked with FAS to develop potential solutions in various realms: infrastructure and the built environment, workforce safety and development, public health, food security and resilience, emergency planning and response, and data indices. Review ideas to combat extreme heat here.
- Improved Accuracy in ICD-10 Coding: Healthcare providers consistently apply accurate ICD-10 coding for extreme heat-related health cases.
- Enhanced Healthcare Provider Knowledge: Healthcare professionals possess comprehensive knowledge on extreme heat-related health impacts, improving patient care and response strategies.
- Strengthened Public Health Response: A coordinated effort results in a more effective and equitable public health response to extreme heat events, reducing health disparities.
- Improved Public Health Resilience:
- Short-Term Outcome: Healthcare providers, armed with enhanced knowledge and training, respond more effectively to extreme heat-related health cases.
- Long-Term Outcome: Reduced morbidity and mortality rates associated with extreme heat events lead to a more resilient and prepared public health system.
- Enhanced Data Accuracy and Surveillance:
- Short-Term Outcome: Improved accuracy in ICD-10 coding facilitates more precise tracking and surveillance of extreme heat-related health outcomes.
- Long-Term Outcome: Comprehensive and accurate data contribute to better-informed public health policies, targeted interventions, and long-term trend analysis.
- Reduced Health Disparities:
- Short-Term Outcome: Incentives and education programs ensure that healthcare providers prioritize accurate coding, reducing disparities in the diagnosis and treatment of extreme heat-related illnesses.
- Long-Term Outcome: Health outcomes become more equitable across diverse populations, mitigating the disproportionate impact of extreme heat on vulnerable communities.
- Increased Public Awareness and Education:
- Short-Term Outcome: Public health campaigns and educational initiatives raise awareness about the health risks associated with extreme heat events.
- Long-Term Outcome: Informed communities adopt preventive measures, reducing the overall burden on healthcare systems and fostering a culture of proactive health management.
- Streamlined Emergency Response and Preparedness:
- Short-Term Outcome: Integrating extreme heat preparedness into emergency response plans results in more efficient and coordinated efforts during heatwaves.
- Long-Term Outcome: Improved community resilience, reduced strain on emergency services, and better protection for vulnerable populations during extreme heat events.
- Increased Collaboration Across Agencies:
- Short-Term Outcome: Collaborative efforts between OCCHE, CMS, HRSA, AHRQ, FEMA, DoD, and the Department of the Interior result in streamlined information sharing and joint initiatives.
- Long-Term Outcome: Enhanced cross-agency collaboration establishes a model for addressing complex public health challenges, fostering a more integrated and responsive government approach.
- Empowered Healthcare Workforce:
- Short-Term Outcome: Incentives for accurate coding and targeted education empower healthcare professionals to address the unique challenges posed by extreme heat.
- Long-Term Outcome: A more resilient and adaptive healthcare workforce is equipped to handle emerging health threats, contributing to overall workforce well-being and satisfaction.
- Informed Policy Decision-Making:
- Short-Term Outcome: Policymakers utilize accurate data and insights to make informed decisions related to extreme heat adaptation and mitigation strategies.
- Long-Term Outcome: The integration of health data into broader climate and policy discussions leads to more effective, evidence-based policies at local, regional, and national levels.