Disaster Policy Nerds Explain the Good, Bad, and Ugly in FEMA Review Council Report
It’s here! After months of delay, the council tasked by President Trump to review the Federal Emergency Management Agency (FEMA) released its final report earlier this month.
If you’re not a disaster policy nerd like we are, here’s some quick background.
Up until the founding of FEMA in 1979 under President Jimmy Carter, disaster response in the United States was largely disorganized and reactive. The agency has since gone through several major updates. Passage of the Robert T. Stafford Act in 1988 established the formal mechanism for disaster declarations and federal disaster response, while in the years following the 9/11 attacks FEMA transitioned from an independent cabinet agency to part of the newly established Department of Homeland Security.
Recently, FEMA has come under intense scrutiny from the second Trump administration for being seen as ineffective, bureaucratic, and in some cases politically biased against him. While FEMA has had issues in the past related to delayed response times and survivors receiving aid (and criticism with how it handled Hurricane Maria), reports show that many of these issues may be related to an increase in major disasters due to climate change, as well as a lack of regular training, sufficient funding, and adequate staffing – rather than structural issues with the agency. In addition, traditionally “red” states (like Texas, Louisiana, and Florida) typically receive more FEMA funding due to the amount of disasters they experience, so claims of political bias are largely unfounded.
Yet when Trump took office for the second time, there were calls to get rid of FEMA altogether. However, after pushback from citizens and lawmakers and several major disasters, the aforementioned council has opted to avoid recommending completely dismantling the agency. Instead, the council proposes major changes to the way FEMA operates (the council repeatedly refers to a “transformed agency”), via ten general recommendations. Here’s our quick snapshot of the good, the bad, and the ugly of these recommendations, with more detail below:
- The Good: An emphasis on mitigation and streamlining the application process for disaster survivors, along with a focus on getting money to states and survivors quicker.
- The Bad: Shrinking and privatizing most of the National Flood Insurance Program (NFIP), while slashing overall federal funding for disaster response and recovery.
- The Ugly: An unrealistically short timeline for implementing recommendations in the report, as well as a reductionist approach to how disasters enact damage.
Also, a quick note on the United States’ approach to disasters: there is generally an overemphasis on acute economic impacts, and not what they do to systems as a whole long-term. As many disaster researchers will tell you, while hazards can be natural, disasters are not. Disasters are the result of hazards adversely impacting people and their communities due to decisions that increase vulnerability and risk exposure. If we limit our approach to disaster recovery to include only economies and infrastructure, we’ll tend to overlook other critical factors, like public health, social connection, and community wellbeing, that contribute to these vulnerabilities. Just because a house has been rebuilt or power has been restored does not mean recovery has been achieved. Before we implement sweeping changes to the agency responsible for disaster response, it’s important that we as a nation consider this in our approach to disasters.
With that aside, onto the deeper dive into the good, the bad, and the ugly of the review council’s report.
The Good
First, the council re-emphasizes FEMA’s core mission of “[reducing] the loss of life and property and [protecting] the Nation from all hazards”, with the guiding principle of disaster response being “locally executed, state or tribally managed, and federally supported”. This is in line with the survivor-led response approach that many local recovery groups have recommended. Communities have the local knowledge and boots-on-the-ground presence needed to ensure that recovery efforts are appropriate for their situations and contexts, but often lack the funding to implement tailored solutions. The council suggests that FEMA strengthen regional coordination, which could in turn support community- and survivor-led response.
Another positive is the council’s emphasis on rapid mitigation and hardening support to increase efficiency and prevent damage from future disasters. This includes modernizing the federal disaster response by implementing the National Resilience Strategy and updating flood risk information and land use to prevent building in flood plains, both common-sense solutions that can prevent the worst damage from disasters before they even occur. They additionally recommend a two-phase program that would replace the Hazard Grant Mitigation Program (HGMP) with a new program designed to more rapidly distribute federal funding to states (the first 5% of federal funds within the first 30 days following a disaster declaration, followed by an additional 10% within six months). The council calls this new program the “Refined Risk Reduction” Program (R3P), and could potentially address issues survivors have brought up with administrative burden around disaster aid, such as by modifying the Individual Assistance Program and consolidating relief applications into a single direct payment program. There are also specific relief allocations for renters (who often get left out of recovery discussions), including the equivalent of three to six months of rent.
Finally, a recommendation could be either positive or negative (depending at least partially on the details of implementation) is changing how surviving homeowners get reimbursed for individual assistance, including how the amounts are calculated. Currently, home repair assistance payments are capped at $25,000 and based on loss estimates, regardless of property value. The council suggests changing this cap to no more than 15% of the home valuation (so a home valued at $250,000 would qualify for a maximum payment of $37,500), but expanding the purpose of such payments to cover everything from home repairs to funeral costs – i.e., requiring the payments to stretch further than they do currently. Another issue is that FEMA funding has been found to favor wealthier individuals, and basing funding on home valuation has the potential to further drive disparities in aid. Supplemental funding opportunities and/or proactive aid and assistance for lower-income families could potentially reduce this risk.
The Bad
Perhaps the most concerning recommendation of the council is its call for a “lean FEMA workforce”. While the council is less aggressive overall in demanding FEMA staffing reductions than previous drafts (and now just calls for a strategic review of requirements to “determine appropriate staffing levels”), further staffing reductions could exacerbate issues we’re already seeing with FEMA from previous personnel cuts. Disaster survivors have also condemned further FEMA staffing cuts. The council also suggests adjusting how insurance rates are calculated under the National Flood Insurance Program (NFIP) and shifting more flood insurance policies to private markets, which could prohibitively increase premiums, decrease regulation, and lead to more uninsurance and underinsurance in risk-prone areas unless there are appropriate safety measures in place.
Finally, the council recommends decreasing the overall federal share of disaster assistance funding from 75–100% to 50–75% of costs, with states expected to cover the rest. Many states do not have resources to cover the difference – at least in the near term. With a sufficient transition period, though, more heavily weighting state responsibility for disaster aid may increase sustainability in the long term given that the Disaster Relief Fund repeatedly runs low on funds, and that there are concerns with fund depletion as disasters continue to increase in frequency and severity. Another concern is the council’s prioritization of “high performing states”. While this would encourage more states to have hazard mitigation plans in place, it could result in biased decision-making that would favor certain states, and may leave states with fewer financial resources or rare disaster occurrence with less support when it’s most needed.
The Ugly
The biggest overall issue with the council’s suggestions is that they recommend a 2–3 year timeline for states and tribal governments to prepare their fiscal and physical resources to lead disaster response efforts (rather than relying on FEMA). This is an unrealistic timeline, as many states do not currently have sufficient emergency management resources, legislation to establish and support relief funding, or identified revenue streams to pay for the increased cost-share for states. And on top of that, some state governments (like Texas) only meet every two years, making the 2-3 year timeline impossible. Another issue with the council is the apparent bias in its makeup. While it did include representatives with leadership and emergency management experience from hard-hit states like Florida, Texas, Louisiana, Mississippi, and Virginia, there was a notable lack of members from other disaster-prone parts of the country, like California.
Another problem is the council’s recommendation of using a parametric insurance program to replace FEMA’s current Public Assistance Program (the main funding source for community-level disaster recovery). Parametric insurance is a type of insurance where payments are disbursed almost immediately following certain trigger events, and while it has demonstrated potential in rapidly distributing funds after certain hazardous events, there are too many variables to feasibly consider replacing the entire Public Assistance Program in 2-3 years. For example, the council includes an example of determining payment amounts by hurricane category (e.g.,: a Category 2 hurricane would disburse less funds than a Category 4). However, hurricane categories are based on wind speed alone, and hurricanes with weaker winds can still do extensive damage through other means, like storm surge and rainfall (Hurricane Ike in 2008 and Hurricane Harvey in 2017 are two such examples). These complexities would have to be accounted for when establishing the thresholds of a parametric insurance framework, and without rigorous pilot testing, runs the risk of over or underpaying states following disasters.
One final concern is the council made no mention whatsoever of the BRIC (Building Resilient Infrastructure and Communities) grant program. This absence from the report likely means BRIC is not a priority for current leadership, despite it being one of the largest sources for proactive mitigation funding for states and communities. BRIC has been the subject of much consternation following its abrupt cancellation in April of 2025 and its later reinstatement in March of 2026 (following a lawsuit from several states). However, there is now a heavy focus on “shovel-ready projects” (i.e. physical infrastructure projects that have already been planned out). While this sounds good for efficiency, as we noted earlier, not all infrastructure critical to community wellbeing and recovery is physical and “shovel-ready”. Things like social and public health infrastructure are just as important for disaster recovery, but tend to be overlooked in recovery efforts. In limiting BRIC funding to these types of projects, states and local governments will be unable to be truly proactive in their mitigation efforts to prevent future damage from disasters.
Conclusion
The review council’s recommendations are not as bad as they could have been and FEMA’s continued existence seems to be safe (for now). Indeed, many of the recommendations could yield positive results, especially when it comes to reducing the burden and obstacles that survivors face in getting help. However, some of the recommendations (like using parametric insurance methods, reducing state assistance, and attempting to implement sweeping changes over a fast 2–3 year timeline) could pose problems for states, communities, and survivors, and a longer transition period with pilot testing will be needed to ensure these changes happen efficiently and effectively.
Long-term effects of disasters: an ongoing threat to public health
2025 was a costly extreme weather year. January kicked off with the Palisades and Eaton wildfires, which caused an estimated 31 deaths in Los Angeles County and billions of dollars in economic damages. Texas experienced some of its worst flooding in decades in July, resulting in the deaths of more than 100 people, many of whom were children staying at an overnight summer camp. In October, flooding in Alaska led to at least one person’s death and the displacement of more than 1,500 others as whole villages were inundated from the remnants of Typhoon Halong.
However, even though 2025 is over, the effects of these disasters are not.
There is considerable interest from both sides of the political aisle in reforming FEMA. The reform conversation has largely focused on where responsibility for disaster response should sit – with the federal government, or with the states? But from a public health perspective, we should be talking about something even more important: our hyperfocus on short-term disaster effects that causes us to neglect the longer-term needs of disaster-affected survivors and communities.
Disasters create ripple effects on health that can extend into the months, years, and even decades after fires are put out, winds die down, and floodwaters recede. While these effects are hard to precisely measure, they are undeniably potent. Official data, for instance, indicate that hurricanes and other tropical cyclones cause an average of 24 deaths per storm. That number is too high – every death is a tragedy – but it pales in comparison to the 7,000 – 11,000 deaths that studies estimate actually come from storms’ longer-term consequences.
With extreme weather events becoming ever-more common, there is a national and moral imperative to rethink not just who responds to disasters, but for how long and to what end. This issue brief presents an overview of the ways in which disasters affect public health and well-being in the long term, as well as suggestions for disaster governance reform viewed through a public health lens.
Disasters impose sustained effects on physical and mental health
The evidence is clear: disasters cause suffering that persists long after national attention turns elsewhere. The same study cited above also found that the adverse health effects of hurricanes and other tropical cyclones were most pronounced in infants under the age of one almost two years after a given storm. If you do the math, this means the storms continued to increase the infants’ risk of death despite them not having even been conceived at landfall, suggesting that the true damage from hurricanes (and likely other natural hazards) comes from the stress they put on families and communities. This doesn’t just apply to infants, but other vulnerable groups, such as older adults. For example, older adults who lived through Superstorm Sandy had higher risks of cardiovascular disease and all-cause mortality five years after the storm had passed.
Why do exposures to extreme weather events have these long-term health impacts? More research is needed, but so far at least three likely pathways have been identified.
Disasters can expose survivors to hazardous conditions that increase their risk of chronic illnesses. For example, hazardous particles from wildfire smoke can settle into people’s lungs and negatively affect lung function for years, which is especially dangerous when individuals have pre-existing respiratory conditions like asthma or COPD. This decreased lung function has been shown to last at least two years after the wildfire for some individuals. Individuals living in high-risk areas where wildfires frequently occur may never get the chance to fully recover.
The physical damages from a disaster to the built and natural environments that surround survivors can become hazardous to human health. For instance, when disasters knock out power during a heatwave or cold snap, individuals can be exposed to dangerous temperature extremes. Heatwaves alone can increase the risk of chronic kidney disease, and can even make the body age faster, while those who have experienced heat illness and heat stroke are at risk of organ damage, including damage to the brain. Other disasters can spread harmful toxins. Floodwaters often contain hazardous waste from runoff and sewage system overflow, increasing the risk of illness in the months following the flood. Homes and businesses that flooded are also at risk of growing toxic mold, especially in warm and humid environments (like the hurricane-prone Southeast United States). What is especially challenging about mold is that it can be difficult to detect and can persist for years unless treated, leading to families living in toxic environments without ever knowing it. Exposure to mold increases the risk for various diseases and health problems, including asthma attacks and infections, and are particularly concerning for sensitive individuals, like those who are immunocompromised. Similarly, when wildfires burn houses, vehicles, and other infrastructure, this releases toxic ash and other debris into the air for hundreds of miles, contaminating the lands and waters of communities for years. Airborne toxins released during wildfires can settle on indoor surfaces and in heating, ventilation, and air conditioning systems. These harmful exposures can contribute to serious long-term health conditions like cardiovascular and respiratory diseases and cancer.
Living through a disaster and then navigating a byzantine recovery landscape can contribute to chronic stress that takes a toll on mental and physical health. Several studies suggest that exposure to disasters increases the risk of mental health challenges like depression, anxiety, and post-traumatic stress, affects people of all age groups, including children, and can last for years. One study found that survivors of Hurricane Katrina were still dealing with post-traumatic stress symptoms twelve years later. Poor mental health can lead to poor physical health in the long term (such as an increased risk of chronic diseases and premature death), as people deal with the toll chronic stress can have on the body. This stress can also increase risk of cognitive decline and dementia, and considering that chronic stress is common among disaster survivors, it’s no wonder that wildfires, hurricanes, and heat waves have all been found to be associated with cognitive decline and dementia too.
Disasters disrupt healthcare delivery and operations
Access to healthcare, including going to doctor’s appointments, getting prescription refills, and receiving specialty treatment (like chemotherapy or dialysis), is critical for keeping people healthy and maintaining quality of life. Unfortunately, disasters often displace people from their homes, communities, and from the healthcare services they depend on. For many people, displacement after a disaster is permanent. These people must then navigate entirely new environments, find new healthcare providers, and become re-established with the medical system all the while securing a place to live, meeting their other needs, and dealing with the stress of losing their life as they knew it. Medical care often falls to the wayside in the chaos of disaster recovery, leaving survivors vulnerable to worsening health conditions over time.

Disasters often displace people from their homes, communities, and from the healthcare services they depend on. Take for instance, these flooded homes adjacent to the Red River in North Dakota, via Wikimedia Commons
Even if displacement is only temporary, when residents return to their homes and communities they may find that their healthcare options no longer exist, as disasters can disrupt or destroy entire healthcare systems. Damaged clinics and hospitals may be temporarily or permanently shut down, creating healthcare “deserts” that lack sufficient healthcare infrastructure to treat people. Rural areas are particularly vulnerable to these disruptions, as they already face limited budgets and fewer resources – leaving less “cushion” to absorb disaster impacts. Disasters can also disrupt supply chains, impacting the quality of medical care for entire regions. This occurred when Hurricane Helene flooded one of the major medical IV suppliers in the United States, leading to IV shortages at healthcare facilities that persisted even months after the hurricane.
Disasters drive housing insecurity
Having an affordable and stable place to live is one of the most important social determinants of health. Access to housing that is safe, clean, and sheltered from the elements can affect everything from risk of hospitalization, the number of medications someone takes, mortality, and overall quality of life. Unfortunately, housing is also one of the most vulnerable and deeply personal domains to be affected by disasters. Many are forced to flee from fires, floods, high winds, or other dangerous conditions presented by natural hazards, and people can be displaced from their homes for months or years at a time – or even permanently. This displacement is associated with numerous mental and physical health issues, including risk of death. And as insurance premiums continue to increase or insurance companies withdraw from states altogether because of the increasing frequency and intensity of disasters, homeowners may be unable to afford property insurance altogether in the near future, further reducing the chances of ever being able to return home.
Even for those who could return home, the cost of recovery may be too prohibitive. For wildfire survivors, many insurance companies do not include smoke damage testing and remediation costs under their policies, and even some plans that cover smoke damage may refuse to remediate, which can cost thousands of dollars out-of-pocket. For flood survivors, mold can be difficult to detect and may require the services of a professional cleaner, which can be prohibitively expensive. Even if someone has flood insurance through the National Flood Insurance Program (NFIP), mold remediation is not typically covered, creating barriers for people trying to make their homes healthy to live in again. If someone’s insurance plan covers mold remediation, many plans make exceptions if action to prevent mold growth is not taken in the immediate days after the flood, which can pose a problem for those blocked from returning under emergency orders. These policy barriers result in survivors having to either relocate away from their homes and communities, or continue to live in unsafe conditions that can chronically affect their health.
Opportunities for Action
Our nation needs policies that understand and address the true public health effects of disasters over the months and years after the disaster is technically “over”. Opportunities for action include:
Update how health impacts are measured. Tracking of health impacts from disasters is generally limited to the direct impacts of disasters (i.e., injuries and deaths, like drownings from floods and smoke asphyxiation from wildfires). Few official data sources capture the indirect effects of disasters that take more time to manifest. Impact assessments should include epidemiological methodology that can capture these indirect effects, such as excess death calculations, to begin to truly understand and quantify the extent of disasters on peoples’ health.
Reinstate disrupted grants and other funding for health research relevant to disasters. Recent budget cuts, terminated grants, and increased hostility towards public health threaten our ability to understand – and therefore effectively address – the health impacts of disasters. Policymakers should reinstate such funding, and look for opportunities to prioritize research related to the longer-term and often overlooked impacts.
Invest in the physical resilience of healthcare infrastructure. Investments in the physical resilience of healthcare infrastructure (such as the installation of hurricane-proof glass, flood barriers, air filtration systems, etc.) strengthens disaster resilience and reduces the overall health impacts of disasters in the short and long terms. Studies show that these investments pay off in the long run, with every dollar dedicated to resilience yielding multiple dollars in avoided societal costs – and providing strong justification for state and federal resources to catalyze and support such investments.
Strengthen medical supply chains. The Strategic National Stockpile (SNS) provides a critical reserve of essential medical equipment, buffering health supply chains against disruptions from events like disasters. Congress should act to increase the SNS, particularly in locations that could provide rapid response of medical provisions to disaster-affected areas, and ensure that key medical suppliers are adequately prepared for future disasters.
Help with housing. One of the most efficient and effective means of disaster recovery is ensuring survivors have access to stable, safe, and healthy housing. One way to achieve this is by creating a centralized agency or entity (such as the Joint County-State Housing Task Force established in the wake of the 2025 Los Angeles wildfires) to handle housing-specific needs from disasters and for those displaced. At the federal level, this could look like integration of housing-relevant capabilities and authorities across agencies such as the Federal Emergency Management Administration (FEMA, particularly FEMA’s National Flood Insurance Program), the Department of Housing and Urban Development, and the Small Business Administration. Other options could include streamlining access to housing relief funds (such as by creating a consolidated hub for applications for assistance), creating pre-approved resilient home designs that can be fast-tracked for permitting and construction, and establishing well-defined responsibilities for remediation efforts, including cleaning of contaminated lands and homes. (Note: many of these issues are addressed in the proposed FEMA Act of 2025.)
Support on-the-ground efforts. Federal and state agencies can support nonprofit rapid response teams (such as SBP) equipped with cleaning and rebuilding supplies. These teams can be rapidly organized and deployed to mitigate physical damage from disasters, making it faster and safer for survivors to return home. As these teams often include members of affected communities, they often have a deep understanding of what the needs of survivors truly are, enabling more efficient use of resources.
Conclusion
Disaster survivors want reform – but drastically reducing FEMA’s workforce without investing in any new disaster-response capabilities isn’t the type of reform they want. Rather, survivors are looking for leaders to institute more holistic disaster-governance strategies that include efforts to minimize long-term negative impacts, instead of the drop-in/rapid withdrawal pattern historically demonstrated. If we are to make (and keep) Americans healthy, then it’s time to make sure considerations for the long-term health needs of disaster survivors are being met, even after the winds, floods, and fires are gone.
A National Blueprint for Whole Health Transformation
Despite spending over 17% of GDP on health care, Americans live shorter and less healthy lives than their peers in other high-income countries. Rising chronic disease and mental health challenges as well as clinician burnout expose the limits of a system built to treat illness rather than create health. Addressing chronic disease while controlling healthcare costs is a bipartisan goal, the question now is how to achieve this shared goal? A policy window is opening now as Congress debates health care again – and in our view, it’s time for a “whole health” upgrade.
Whole Health is a proven, evidence-based framework that integrates medical care, behavioral health, public health, and community support so that people can live healthier, longer, and more meaningful lives. Pioneered by the Veterans Health Administration, Whole Health offers a redesign to U.S. health and social systems: it organizes how health is created and supported across sectors, shifting power and responsibility from institutions to people and communities. It begins with what matters most to people–their purpose, aspirations, and connections–and aligns prevention, clinical care, and social supports accordingly. Treating Whole Health as a shared public priority would help ensure that every community has the conditions to thrive.
Challenge and Opportunity
The U.S. health system spends over $4 trillion annually, more per capita than any other nation, yet underperforms on life expectancy, infant mortality, and chronic disease management. The prevailing fee-for-service model fragments care across medical, behavioral, and social domains, rewarding treatment over prevention. This fragmentation drives costs upward, fuels clinician burnout, and leaves many communities without coordinated support.
At this inflection point in our declining health outcomes and growing public awareness of the failures of our health system, federal prevention and public health programs are under review, governors are seeking cost-effective chronic disease solutions, and the National Academies is advocating for new healthcare models. Additionally, public demand for evidence-based well-being is growing, with 65% of Americans prioritizing mental and social health. There is clear demand for transformation in our health care system to deliver results in a much more efficient and cost effective way.
Veterans Health Administration’s Whole Health System Debuted in 2011
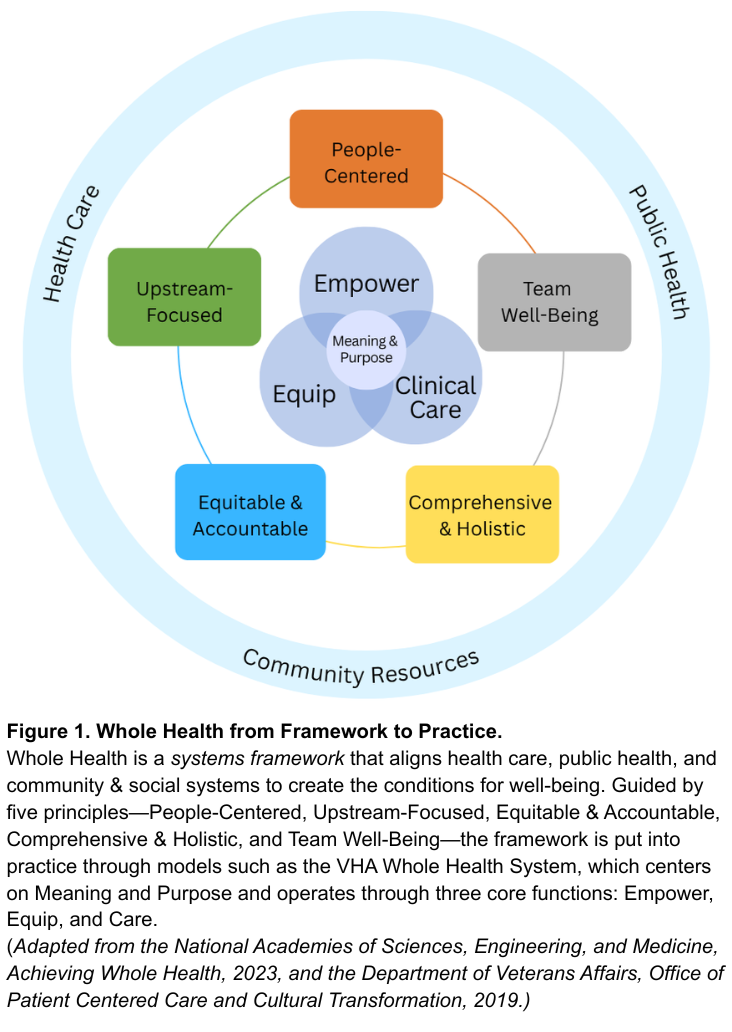
Whole Health offers a system-wide redesign for the challenge at hand. As defined by the National Academies of Sciences, Engineering, and Medicine, Whole Health is a framework for organizing how health is created and supported across sectors. It integrates medical care, behavioral health, public health, and community resources. As shown in Figure 1, the framework connects five system principles—People-Centered, Upstream-Focused, Equitable & Accountable, Comprehensive & Holistic, and Team Well-Being–that guide implementation across health and social support systems. The nation’s largest health system, the Veterans Health Administration’s (VHA), has demonstrated this framework in clinical practice through their Whole Health System since 2011. The VHA’s Whole Health System operates through three core functions: Empower (helping individuals define purpose), Equip (providing community resources like peer support), and Clinical Care (delivering coordinated, team-based care). Together, these elements align with what matters most to people, shifting the locus of control from expert-driven systems to shared agency through partnerships. The Whole Health System at the VHA has reduced opioid use and improved chronic disease outcomes.
Successful State Examples
Beyond the VHA, states have also demonstrated the possibility and benefits of Whole Health models. North Carolina’s Healthy Opportunities Pilots extended Medicaid coverage to housing, food, and transportation, showing fewer emergency visits and savings of about $85 per member per month. Vermont’s Blueprint for Health links primary care practices with community health teams and social services, reducing expenditures by about $480 per person annually and boosting preventive screenings. Finally, the Program of All-Inclusive Care for the Elderly (PACE), currently being implemented in 33 states, utilizes both Medicare and Medicaid funding to coordinate medical and social care for older adults with complex medical needs. While improvements can be made to national program-wide evaluation, states like Kansas have done evaluations that have found that the PACE program is less expensive than nursing homes per beneficiary and that nursing home admissions decline by 5% to 15% for beneficiaries.
Success across each of these examples relies on three pillars: (1) integrating medical, behavioral, social, and public health resources; (2) sustainable financing that prioritizes prevention and coordination; and (3) rigorous evaluation of outcomes that matter to people and communities. While these programs are early signs of success of Whole Health models, without coordinated leadership, efforts will fragment into isolated pilots and it will be challenging to learn and evolve.
A policy window for rethinking the health care system is opening. At this national inflection point, the U.S. can work to build a unified Whole Health strategy that enables a more effective, affordable and resilient health system.
Plan of Action
To act on this opportunity, federal and state leaders can take the following coordinated actions to embed Whole Health as a unifying framework across health, social, and wellbeing systems.
Recommendation 1. Declare Whole Health a Federal and State Priority.
Whole Health should become a unifying value across federal and state government action on health and wellbeing, embedding prevention, connection, and integration into how health and social systems are organized, financed, and delivered. Actions include:
- Federal Executive Action. The Executive Office of the President should create a Whole Health Strategic Council that brings together Veterans Affairs (VA), Health and Human Services (e.g. Centers for Disease Control and Prevention, Centers for Medicare and Medicaid (CMS), and Health Resources and Services Administration (HRSA)), Housing and Urban Development (HUD), and the U.S. Department of Agriculture (USDA) to align strategies, budgets, and programs with Whole Health principles through cross-agency guidance and joint planning. This council should also work with Governors to establish evidence-based benchmarks for Whole Health operations and evaluation (e.g., person-centered planning, peer support, team integration) and shared outcome metrics for well-being and population health.
- U.S. Congressional Action. Authorize whole health benefits, like housing assistance, nutrition counseling, transportation to appointments, peer support programs, and well-being centers as reimbursable services under Medicare, Medicaid and the Affordable Care Act health subsidies.
- State Action. Adopt Whole Health models through Medicaid managed-care contracts and through CDC and HRSA grant implementation. States should also develop support for Whole Health services in trusted local settings such as libraries, faith-based organizations, senior centers, to reach people where they live and gather.
Recommendation 2. Realign Financing and Payment to Reward Prevention and Team-Based Care.
Federal payment modalities need to shift from a fee-for-service model toward hybrid value-based models. Models such as per-member-per-month payments with quality incentives, can sustain comprehensive, team-based care while delivering outcomes that matter, like reductions in chronic disease and overall perceived wellbeing. Actions include:
- Federal Executive Action. Expand Advanced Primary Care Management (APCM) payments to include Whole Health teams, including clinicians, peer coaches, and community health workers. Ensure that this funding supports coordination, person-centered planning, and upstream prevention, such as food as medicine programs. Further, CMS can expand reimbursements to community health workers and peer support roles and standardize their scope-of-practice rules across states.
- U.S. Congressional Action. Invest in Medicare and Medicaid innovation programs, such as the CMS Innovation Center (CMMI), that reward prevention and chronic disease reduction. Additionally, expand tools for payment flexibility, through Medicaid waivers and state innovation funds, to help states adapt Whole Health models to local needs.
- State Action. Require Medicaid managed-care contracts to reimburse Whole Health services, particularly in underserved and rural areas, and encourage payers to align benefit designs and performance measures around well-being. States should also leverage their state insurance departments to guide and incentivize private health insurers to adopt Whole Health payment models.
Recommendation 3. Strengthen and Expand the Whole Health Workforce.
Whole Health practice needs a broad team to be successful: clinicians, community health workers, peer coaches, community organizations, nutritionists, and educators. To build this workforce, governments need to modernize training, assess the workforce and workplace quality, and connect the fast-growing well-being sector with health and community systems. Actions include:
- Federal Executive Action. Through VA and HRSA establish Whole Health Workforce Centers of Excellence to develop national curricula, set standards, and disseminate evidence on effective Whole Health team-building. Further, CMS should track workforce outcomes such as retention, burnout, and team integration, and evaluate the benefits for health professionals working in Whole Health systems versus traditional health systems.
- U.S. Congressional Action. Expand CMS Graduate Medical Education Funds and HRSA workforce programs to support Whole Health training, certifications, and placements across clinical and community settings.
- State Action. As a part of initiatives to grow the health workforce, state governments should expand the definition of a “health professional” to include Whole Health practitioners. Further, states can leverage their role as a licensure for professionals by creating a “whole health” licensing process that recognizes professionals that meet evidence-based standards for Whole Health.
Recommendation 4. Build a National Learning and Research Infrastructure.
Whole Health programs across the country are proving effective, but lessons remain siloed. A coordinated national system should link evidence, evaluation, and implementation so that successful models can scale quickly and sustainably.
- Federal Executive Action. Direct the Agency for Healthcare Research and Quality, National Institutes of Health, and partner agencies (VA, HUD, USDA) to run pragmatic trials and cost-effectiveness studies of Whole Health interventions that measure well-being across clinical, biomedical, behavioral, and social domains. The federal government should also embed Whole Health frameworks into government-wide research agendas to sustain a culture of evidence-based improvement.
- U.S. Congressional Action. Charter a quasi-governmental entity, modeled on Patient-Centered Outcomes Research Institute (PCORI), to coordinate Whole Health demonstration sites and research. This new entity should partner with CMMI, HRSA and VA to test Whole Health payment and delivery models under real-world conditions. This entity should also establish an interagency team as well as state network to address payment, regulatory, and privacy barriers identified by sites and pilots.
- State Action. Partner with federal agencies through innovation waivers (e.g. 1115 waivers and 1332 waivers) and learning collaboratives to test Whole Health models and share data across state systems and with the federal government.
Conclusion
The United States spends more on health care than any other nation yet delivers poorer outcomes. Whole Health offers a proven path to reverse this trend, reframing care around prevention, purpose, and integration across health and social systems. Embedding Whole Health as the operating system for America’s health requires three shifts: (1) redefining the purpose from treating disease to optimizing health and well-being; (2) restructuring care to empower, equip, and treat through team-based and community-linked approaches; and (3) rebalancing control from expert-driven systems to partnerships guided by what matters most to people and communities. Federal and state leaders have the opportunity to turn scattered Whole Health pilots to a coordinated national strategy. The cost of inaction is continued fragmentation; the reward of action is a healthier and more resilient nation.
This memo produced as part of Strengthening Pathways to Disease Prevention and Improved Health Outcomes.
Both approaches emphasize caring for people as integrated beings rather than as a collection of diseases, but they differ in scope and application. Whole Person Health, as used by NIH, focuses on the biological, psychological, and behavioral systems within an individual—it is primarily a research framework for understanding health across body systems. Whole Health is a systems framework that extends beyond the individual to include families, communities, and environments. It integrates medical care, behavioral health, public health, and social support around what matters most to each person. In short, Whole Person Health is about how the body and mind work together; Whole Health is about how health, social, and community systems work together to create the conditions for well-being. Policymakers can use Whole Health to guide financing, workforce, and infrastructure reforms that translate Whole Person Health science into everyday practice.
Integrative Health combines evidence-based conventional and complementary approaches such as mindfulness, acupuncture, yoga, and nutrition to support healing of the whole person. Whole Health extends further. It includes prevention, self-care, and personal agency, and moves beyond the clinic to connect medical care with social, behavioral, and community dimensions of health. Whole Health uses integrative approaches when evidence supports them, but it is ultimately a systems model that aligns health, social, and community supports around what matters most to people. For policymakers, it provides a structure for integrating clinical and community services within financing and workforce strategies.
They share a common foundation but differ in scope and audience. The VA Whole Health System, developed by the Department of Veterans Affairs, is an operational model, a way of delivering care that helps veterans identify what matters most, supports self-care and skill building, and provides team-based clinical treatment. The National Academies’ Whole Health framework builds on the VA’s experience and expands it to the national level. It is a policy and systems framework that applies Whole Health principles across all populations and connects health care with public health, behavioral health, and community systems. In short, the VA model shows how Whole Health works in practice, while the National Academies framework shows how it can guide national policy and system alignment.
Poison in our Communities: Impacts of the Nuclear Weapons Industry across America
In 1942, the United States formally began the Manhattan Project, which led to the production, testing, and use of nuclear weapons. In August 1945, the United States dropped two nuclear weapons on Japanese cities, killing around 200,000 people by the end of 1945 and leaving survivors with cancer, leukemia and other illnesses caused by radiation exposure. While this was their only use in wartime, states have detonated nuclear weapons many times since for testing purposes, producing radioactive fallout. Many U.S. nuclear weapons production activities, including the mining of uranium and testing of the weapons themselves, have occurred outside of the continental United States. Notably, explosive testing in the Pacific islands and ocean spread radioactive fallout to Marshallese, Japanese, and Gilbertese people, forcibly displacing entire communities and producing intergenerational illnesses.
Much of the scholarship surrounding the effects of nuclear weapons on environmental and human health is framed within a potential detonation scenario. For example, studies have shown that even a regional nuclear war would cause millions of immediate deaths and trigger a “nuclear winter,” a shift in the climate that would disrupt agricultural production, thus killing hundreds of millions more through starvation. Additionally, in 2024, the United Nations General Assembly voted to create an independent scientific panel to study the health, environmental and economic consequences of nuclear war. While such research is crucial for understanding the consequences of nuclear weapons use, nuclear weapons are built, maintained, and deployed everyday, impacting communities at every stage even before detonation. Studying only the predictive futures of the use of a nuclear weapon in war is insufficient in understanding nuclear weapons’ holistic humanitarian impact. According to former Secretary of Defense Lloyd J. Austin III, “The heart of American deterrence is the people who protect us and our allies. Here at STRATCOM, you proudly stand up—day in and day out and around the clock—to defend us from catastrophe and to build a safer and more peaceful future. So let us always ensure that the most dangerous weapons ever produced by human science are managed with the greatest responsibility ever produced by human government.” Nuclear deterrence theory contends that a retaliatory nuclear strike is so threatening that an adversary will not attack in the first place. Thus, nuclear advocates often suggest that these weapons protect American citizens and the U.S. homeland. This report demonstrates, however, that the creation and sustainment of the nuclear deterrent harms members of the American public. As the United States continues nuclear modernization on all legs of its nuclear triad through the creation of new variants of warheads, missiles, and delivery platforms, examining the effects of nuclear weapons production on the public is ever more pressing.
Impacts of Extreme Heat on Labor
Extreme heat is a major occupational hazard with far-reaching impacts on the national economy as well as worker health and safety. Extreme heat costs an estimated $100 billion per year in lost productivity, and causes an average of at least 3,389 heat-related injuries and 33 heat-related fatalities annually – numbers that are likely vast undercounts. To protect workers, Congress must mandate a federal heat standard, retain federal workers with expertise in heat stress management strategies, and establish Centers of Excellence to support research, training, and sector-specific mitigation strategies. Through investments in infrastructure for heat safety, Congress can save lives, protect the economy, and enhance resilience nationwide.
Heat-Related Risks are Heightened in Many Work Environments
Extreme heat puts workers of all types at risk: OSHA has documented hospitalizations and heat-related deaths in close to 275 industries. Some work environments present extreme heat risk, particularly those involving high exposures to the outdoors and limited access to cooling. With roughly one in three U.S. employees regularly working outdoors, a large share of the workforce is at elevated risk during summer months. Indoor workers also face high exposure, especially in kitchens, warehouses, manufacturing plants, and other poorly ventilated environments because heat and humidity easily build up in enclosed spaces without adequate air flow and climate-control.
Business and Economic Impacts of High Heat Exposure in the Workplace
On top of the $100 billion in direct annual losses, high temperatures are also linked to increased healthcare costs for employers and workers’ compensation claims, with claim frequencies rising by up to 10% during temperature extremes. Some industries are more exposed than others; for example, agriculture, construction, and utility companies face twice the risk of incurring increased healthcare claims due to extreme weather and other environmental conditions. This growing number of claims increases companies’ experience modification rates, which insurers use as a key factor for calculating higher future premiums. Higher premiums translate to greater insurance and overall operating costs, which is especially burdensome for small and low-margin businesses. Despite all these risks, many employers continue to underestimate the financial burden of extreme heat and other weather-related health impacts.
Many Military Personnel and Federal Workers Face Above-Average Risks of Heat-Related Illness
Military personnel, federal law enforcement officers, border patrol officers, wildland firefighters, federal transportation workers like railroad inspectors, and postal employees are all in positions that require long, labor-intensive hours outdoors, raising the risk for heat-related illness. In 2024, heat-related illnesses were among the top five most reported medical events among U.S. active duty service members. Without consistent standards in place to protect these workers from extreme heat, military and other federal operations will continue to be vulnerable to disruption and reduced workforce capacity.
Advancing Solutions: Establish a Strong Federal Heat Standard and Sector-Specific Centers of Excellence for Heat Workplace Safety
To begin to address heat-related injuries and illnesses in workplaces, OSHA in 2022 established the National Emphasis Program (NEP) on Outdoor and Indoor Heat-Related Hazards, which remains in effect until April 2026. As of 2025, OSHA reports that this NEP has conducted nearly 7,000 inspections connected to heat risks, which lead to 60 heat citations and nearly 1,400 “hazard alert” letters being sent to employers.
However, in the absence of a federal mandate for effective heat safety practices, most workplaces rely on voluntary guidance that is not tailored to specific job conditions, backed by consistent data, or subject to enforcement. This puts both workers and businesses at risk. OSHA’s proposed Heat Injury and Illness Prevention rule would be a critical step forward to establishing common-sense baseline protections. According to the agency’s projections, compliance with this standard could prevent thousands of heat-related illnesses and deaths. The projected benefits from reduced fatalities, illness, and injury amount to $9.18 billion per year. Importantly, this action has broad public backing: 90% of American voters support the implementation of federal protections from extreme heat in the workplace.
Congress should act swiftly to ensure OSHA finalizes and enforces a strong, evidence-based heat standard. To do this effectively, it is essential that funding for experts at the National Institute for Occupational Safety and Health (NIOSH) is retained in the FY26 budget request, as these critical workers develop criteria for recommended standards on occupational heat stress. These experts have been impacted by reductions in force at NIOSH, and as of July 2025 have not been brought back by the agency.
Some employers have raised concerns about the technical and financial feasibility of the proposed rule. To address these concerns, Congress should pair regulation with practical support by creating federally funded, sector-specific Centers of Excellence (CoEs)for Heat Workplace Safety. These Centers would develop and implement evidence-based solutions tailored to different work environments, such as agriculture and construction. The CoE approach includes comprehensive data collection at worksites that form the basis of occupational safety and health protocols best practices and policies to enhance productivity, prevent injury and illness, and ensure a return on investment. Once strategies are developed, CoEs implement them, track their impact, and work with workers, employers, and cross-sector partners to ensure long-term success.
By leveraging advanced technology, predictive analytics, and continuously updated industry standards, CoEs can help modernize OSHA regulations and make them more aligned with current workplace realities that go beyond simple compliance or post-injury responses. Federal agencies and other industries with sizable workforces that receive government contracts are key places to develop best practices, technologies, and public-private partnerships for these interventions, all while reducing fiscal risk to the federal government.
Advance AI with Cleaner Air and Healthier Outcomes
Artificial intelligence (AI) is transforming industries, driving innovation, and tackling some of the world’s most pressing challenges. Yet while AI has tremendous potential to advance public health, such as supporting epidemiological research and optimizing healthcare resource allocation, the public health burden of AI due to its contribution to air pollutant emissions has been under-examined. Energy-intensive data centers, often paired with diesel backup generators, are rapidly expanding and degrading air quality through emissions of air pollutants. These emissions exacerbate or cause various adverse health outcomes, from asthma to heart attacks and lung cancer, especially among young children and the elderly. Without sufficient clean and stable energy sources, the annual public health burden from data centers in the United States is projected to reach up to $20 billion by 2030, with households in some communities located near power plants supplying data centers, such as those in Mason County, WV, facing over 200 times greater burdens than others.
Federal, state, and local policymakers should act to accelerate the adoption of cleaner and more stable energy sources and address AI’s expansion that aligns innovation with human well-being, advancing the United States’ leadership in AI while ensuring clean air and healthy communities.
Challenge and Opportunity
Forty-six percent of people in the United States breathe unhealthy levels of air pollution. Ambient air pollution, especially fine particulate matter (PM2.5), is linked to 200,000 deaths each year in the United States. Poor air quality remains the nation’s fifth highest mortality risk factor, resulting in a wide range of immediate and severe health issues that include respiratory diseases, cardiovascular conditions, and premature deaths.
Data centers consume vast amounts of electricity to power and cool the servers running AI models and other computing workloads. According to the Lawrence Berkeley National Laboratory, the growing demand for AI is projected to increase the data centers’ share of the nation’s total electricity consumption to as much as 12% by 2028, up from 4.4% in 2023. Without enough sustainable energy sources like nuclear power, the rapid growth of energy-intensive data centers is likely to exacerbate ambient air pollution and its associated public health impacts.
Data centers typically rely on diesel backup generators for uninterrupted operation during power outages. While the total operation time for routine maintenance of backup generators is limited, these generators can create short-term spikes in PM2.5, NOx, and SO2 that go beyond the baseline environmental and health impacts associated with data center electricity consumption. For example, diesel generators emit 200–600 times more NOx than natural gas-fired power plants per unit of electricity produced. Even brief exposure to high-level NOx can aggravate respiratory symptoms and hospitalizations. A recent report to the Governor and General Assembly of Virginia found that backup generators at data centers emitted approximately 7% of the total permitted pollution levels for these generators in 2023. Based on the Environmental Protection Agency’s COBRA modeling tool, the public health cost of these emissions in Virginia is estimated at approximately $200 million, with health impacts extending to neighboring states and reaching as far as Florida. In Memphis, Tennessee, a set of temporary gas turbines powering a large AI data center, which has not undergone a complete permitting process, is estimated to emit up to 2,000 tons of NOx annually. This has raised significant health concerns among local residents and could result in a total public health burden of $160 million annually. These public health concerns coincide with a paradigm shift that favors dirty energy and potentially delays sustainability goals.
In 2023 alone, air pollution attributed to data centers in the United States resulted in an estimated $5 billion in health-related damages, a figure projected to rise up to $20 billion annually by 2030. This projected cost reflects an estimated 1,300 premature deaths in the United States per year by the end of the decade. While communities near data centers and power plants bear the greatest burden, with some households facing over 200 times greater impacts than others, the health impacts of these facilities extend to communities across the nation. The widespread health impacts of data centers further compound the already uneven distribution of environmental costs and water resource stresses imposed by AI data centers across the country.
While essential for mitigating air pollution and public health risks, transitioning AI data centers to cleaner backup fuels and stable energy sources such as nuclear power presents significant implementation hurdles, including lengthy permitting processes. Clean backup generators that match the reliability of diesel remain limited in real-world applications, and multiple key issues must be addressed to fully transition to cleaner and more stable energy.
While it is clear that data centers pose public health risks, comprehensive evaluations of data center air pollution and related public health impacts are essential to grasp the full extent of the harms these centers pose, yet often remain absent from current practices. Washington State conducted a health risk assessment of diesel particulate pollution from multiple data centers in the Quincy area in 2020. However, most states lack similar evaluations for either existing or newly proposed data centers. To safeguard public health, it is essential to establish transparency frameworks, reporting standards, and compliance requirements for data centers, enabling the assessment of PM2.5, NOₓ, SO₂, and other harmful air pollutants, as well as their short- and long-term health impacts. These mechanisms would also equip state and local governments to make informed decisions about where to site AI data center facilities, balancing technological progress with the protection of community health nationwide.
Finally, limited public awareness, insufficient educational outreach, and a lack of comprehensive decision-making processes further obscure the potential health risks data centers pose to public health. Without robust transparency and community engagement mechanisms, communities housing data center facilities are left with little influence or recourse over developments that may significantly affect their health and environment.
Plan of Action
The United States can build AI systems that not only drive innovation but also promote human well-being, delivering lasting health benefits for generations to come. Federal, state, and local policymakers should adopt a multi-pronged approach to address data center expansion with minimal air pollution and public health impacts, as outlined below.
Federal-level Action
Federal agencies play a crucial role in establishing national standards, coordinating cross-state efforts, and leveraging federal resources to model responsible public health stewardship.
Recommendation 1. Incorporate Public Health Benefits to Accelerate Clean and Stable Energy Adoption for AI Data Centers
Congress should direct relevant federal agencies, including the Department of Energy (DOE), the Nuclear Regulatory Commission (NRC), and the Environmental Protection Agency (EPA), to integrate air pollution reduction and the associated public health benefits into efforts to streamline the permitting process for more sustainable energy sources, such as nuclear power, for AI data centers. Simultaneously, federal resources should be expanded to support research, development, and pilot deployment of alternative low-emission fuels for backup generators while ensuring high reliability.
- Public Health Benefit Quantification. Direct the EPA, in coordination with DOE and public health agencies, to develop standardized methods for estimating the public health benefits (e.g., avoided premature deaths, hospital visits, and economic burden) of using cleaner and more stable energy sources for AI data centers. Require lifecycle emissions modeling of energy sources and translate avoided emissions into quantitative health benefits using established tools such as the EPA’s BenMAP. This should:
- Include modeling of air pollution exposure and health outcomes (e.g., using tools like EPA’s COBRA)
- Incorporate cumulative risks from regional electricity generation and local backup generator emissions
- Account for spatial disparities and vulnerable populations (e.g., children, the elderly, and disadvantaged communities)
- Evaluate both short-term (e.g., generator spikes) and long-term (e.g., chronic exposure) health impacts
- Preferential Permitting. Instruct the DOE to prioritize and streamline permitting for cleaner energy projects (e.g., small modular reactors, advanced geothermal) that demonstrate significant air pollution reduction and health benefits in supporting AI data center infrastructures. Develop a Clean AI Permitting Framework that allows project applicants to submit health benefit assessments as part of the permitting package to justify accelerated review timelines.
- Support for Cleaner Backup Systems. Expand DOE and EPA R&D programs to support pilot projects and commercialization pathways for alternative backup generator technologies, including hydrogen combustion systems and long-duration battery storage. Provide tax credits or grants for early adopters of non-diesel backup technologies in AI-related data center facilities.
- Federal Guidance & Training. Provide technical assistance to state and local agencies to implement the protocol, and fund capacity-building efforts in environmental health departments.
Recommendation 2. Establish a Standardized Emissions Reporting Framework for AI Data Centers
Congress should direct the EPA, in coordination with the National Institute of Standards and Technology (NIST), to develop and implement a standardized reporting framework requiring data centers to publicly disclose their emissions of air pollutants, including PM₂.₅, NOₓ, SO₂, and other hazardous air pollutants associated with backup generators and electricity use.
- Multi-Stakeholder Working Group. Task EPA with convening a multi-stakeholder working group, including representatives from NIST, DOE, state regulators, industry, and public health experts, to define the scope, metrics, and methodologies for emissions reporting.
- Standardization. Develop a federal technical standard that specifies:
- Types of air pollutants that should be reported
- Frequency of reporting (e.g., quarterly or annually)
- Facility-specific disclosures (including generator use and power source profiles)
- Geographic resolution of emissions data
- Public access and data transparency protocols
State-level Action
Recommendation 1. State environmental and public health departments should conduct a health impact assessment (HIA) before and after data center construction to evaluate discrepancies between anticipated and actual health impacts for existing and planned data center operations. To maintain and build trust, HIA findings, methodologies, and limitations should be publicly available and accessible to non-technical audiences (including policymakers, local health departments, and community leaders representing impacted residents), thereby enhancing community-informed action and participation. Reports should focus on the disparate impact between rural and urban communities, with particular attention to overburdened communities that have under-resourced health infrastructure. In addition, states should coordinate HIA and share findings to address cross-boundary pollution risks. This includes accounting for nearby communities across state lines, considering that jurisdictional borders should not constrain public health impacts and analysis.
Recommendation 2. State public health departments should establish a state-funded program that offers community education forums for affected residents to express their concerns about how data centers impact them. These programs should emphasize leading outreach, engaging communities, and contributing to qualitative analysis for HIAs. Health impact assessments should be used as a basis for informed community engagement.
Recommendation 3. States should incorporate air pollutant emissions related to data centers into their implementation of the National Ambient Air Quality Standards (NAAQS) and the development of State Implementation Plans (SIPs). This ensures that affected areas can meet standards and maintain their attainment statuses. To support this, states should evaluate the adequacy of existing regulatory monitors in capturing emissions related to data centers and determine whether additional monitoring infrastructure is required.
Local-level Action
Recommendation 1. Local governments should revise zoning regulations to include stricter and more explicit health-based protections to prevent data center clustering in already overburdened communities. Additionally, zoning ordinances should address colocation factors and evaluate potential cumulative health impacts. A prominent example is Fairfax County, Virginia, which updated its zoning ordinance in September 2024 to regulate the proximity of data centers to residential areas, require noise pollution studies prior to construction, and establish size thresholds. These updates were shaped through community engagement and input.
Recommendation 2. Local governments should appoint public health experts to the zoning boards to ensure data center placement decisions reflect community health priorities, thereby increasing public health expert representation on zoning boards.
Conclusion
While AI can revolutionize industries and improve lives, its energy-intensive nature is also degrading air quality through emissions of air pollutants. To mitigate AI’s growing air pollution and public health risks, a comprehensive assessment of AI’s health impact and transitioning AI data centers to cleaner backup fuels and stable energy sources, such as nuclear power, are essential. By adopting more informed and cleaner AI strategies at the federal and state levels, policymakers can mitigate these harms, promote healthier communities, and ensure AI’s expansion aligns with clean air priorities.
This memo is part of our AI & Energy Policy Sprint, a policy project to shape U.S. policy at the critical intersection of AI and energy. Read more about the Policy Sprint and check out the other memos here.
Impacts of Extreme Heat on Rural Communities
46 million rural Americans face mounting risks from temperature extremes that threaten workforce productivity, raise business operational costs, and strain critical public services. Though extreme heat is often portrayed in research and the media as an urban issue, almost every state in the contiguous U.S. has rural communities with above-average rates of vulnerability to extreme heat. To protect rural America, Congress must address extreme heat’s impacts by repairing rural health systems, strengthening the preparedness of rural businesses, and hardening rural energy infrastructure.
Extreme heat exacerbates rural communities’ unique health vulnerabilities
On average, Americans living in rural areas are twice as likely as those in urban areas to have pre-existing health conditions, like heart disease, diabetes, and asthma, that make them more sensitive to heat-related illness and death. Further compounding the risk, rural places also have larger populations of underinsured and uninsured people than urban areas, with 1 in 6 people lacking insurance.
Limited healthcare infrastructure in rural places worsens these vulnerabilities. Rural areas have higher shortages of healthcare professionals who provide primary care, mental health, and dental services than urban areas. Over the last decade, 100 rural hospitals have closed, and hundreds more are vulnerable to closure. Finally, many rural communities do not have public health departments, and those that do are underfunded and understaffed. Because public health systems and healthcare professionals are the first responders to extreme heat, rural residents are severely underprepared.
Congress should provide flexible resources and technical assistance to rural hospitals to prepare for emerging threats like extreme heat. Additionally, Congress should continue to enable the U.S. Department of Agriculture and the Department of Health and Human Services to provide loans or grant assistance to help rural residents retain access to health services and improve the financial position of rural hospitals and clinics. And because Medicaid expansion correlates with better rural hospital financial performance and fewer closures, Congress should invest in Medicaid to protect rural healthcare access.
Extreme heat puts rural businesses and workers at risk
Rural economic health relies on the outdoors (e.g., recreation tourism) and outdoor labor (e.g., agriculture and oil and gas extraction). Extreme heat in many of these places makes it dangerous to be outside, which impacts worker productivity and local business revenues. Indoor workers in facilities like manufacturing plants, food processing, and warehouses also face heat-related safety threats due to the presence of heat-producing machines and poorly ventilated buildings with limited cooling. These facilities are rapidly growing components of rural economies, as these sectors employ almost 1 in 5 rural workers.
Simple protections like water, rest, shade, and cooling can improve productivity and generate returns on investments. But small-to-medium rural enterprises need support to adopt affordable cooling systems, shade and passive cooling infrastructure, and worker safety measures that reduce heat-related disruptions. Congress should help rural businesses reduce heat’s risks by appropriating funding to support workplace heat risk reduction and practical training on worker protections. Additionally, Congress should require OSHA to finalize a federal workplace heat standard.
Extreme heat threatens rural energy security
When a power outage happens during a severe extreme heat event, the chance of heat-related illness and death increases exponentially. Extreme heat strains power infrastructure, increasing the risk of power outages. This risk is particularly acute for rural communities, which have limited resources, older infrastructure, and significantly longer waits to restore power after an outage.
Weatherized housing and indoor infrastructure are one of the key protective factors against extreme heat, especially during outages. Yet rural areas often have a higher proportion of older, substandard homes. Manufactured and mobile homes, for example, compose 15% of the rural housing stock and are the one of the most at-risk housing types for extreme heat exposure. When the power is on, rural residents spend 40% more of their income on their energy bills than their urban counterparts. Rural residents in manufactured housing spend an alarming 75% more. Energy debt can force people to choose between paying for life-saving energy or food and key medications, compounding poverty and health outcomes.
To drive the energy independence and economic resilience of rural America, Congress should support investments in energy-efficient and resilient cooling technologies, weatherized homes, localized energy solutions like microgrids, and grid-enhancing technologies.
Economic Impacts of Extreme Heat: Energy
As temperatures rise, the strain on energy infrastructure escalates, creating vulnerabilities for the efficiency of energy generation, grid transmission, and home cooling, which have significant impacts on businesses, households, and critical services. Without action, energy systems will face growing instability, infrastructure failures will persist, and utility burdens will increase. The combined effects of extreme heat cost our nation over $162 billion in 2024 – equivalent to nearly 1% of the U.S. GDP.
The federal government needs to prepare energy systems and the built environment through strategic investments in energy infrastructure — across energy generation, transmission, and use. Doing so includes ensuring electric grids are prepared for extreme heat by establishing an interagency HeatSmart Grids Initiative to assess the risk of energy system failures during extreme heat and the necessary emergency responses. Congress should retain and expand home energy rebates, tax credits, and the Weatherization Assistance Program (WAP) to enable deep retrofits that prepare homes against power outages and cut cooling costs, along with extending the National Initiative to Advance Building Codes (NIABC) to accelerate state and local adoption of code language for extreme heat adaptation.
Challenge & Opportunity: Grid Security
Extreme Heat Reduces Energy Generation and Transmission Efficiency
During a heatwave, the energy grid faces not only surges in demand but also decreased energy production and reduced transmission efficiency. For instance, turbines can become up to 25% less efficient in high temperatures. Other energy sources are also impacted: solar power, for example, produces less electricity as temperatures rise because high heat slows the flow of electrical current. Additionally, transmission lines lose up to 5.8% of their capacity to carry electricity as temperatures increase, resulting in reliability issues such as rolling blackouts. These combined effects slow down the entire energy cycle, making it harder for the grid to meet growing demand and causing power disruptions.
Rising Demand and Grid Load Increase the Threat of Power Outages
Electric grids are under unprecedented strain as record-high temperatures drive up air conditioning use, increasing energy demand in the summer. Power generation and transmission are impeded when demand outpaces supply, causing communities and businesses to experience blackouts. According to data from the North American Electric Reliability Corporation (NERC), between 2024 and 2028, an alarming 300 million people across the United States could face power outages. Texas, California, the Southwest, New England, and much of the Midwest are among the states and regions most at risk of energy emergencies during extreme conditions, according to 2024 NERC data.
Data center build-out, driven by growing demand for artificial intelligence, cloud services, and big data analytics, further adds stress to the grid. Data centers are estimated to consume 9% of US annual electricity generation by 2030. With up to 40% of data centers’ total yearly energy consumption driven by cooling systems, peak demand during the hottest days of the year puts demand on the U.S. electric grid and increases power outage risk.
Power outages bear significant economic costs and put human lives at severe risk. To put this into perspective, a concurrent heat wave and blackout event in Phoenix, Arizona, could put 1 million residents at high risk of heat-related illness, with more than 50% of the city’s population requiring medical care. As we saw with 2024’s Hurricane Beryl, more than 2 million Texans lost power during a heatwave, resulting in up to $1.3 billion in damages to the electric infrastructure in the Houston area and significant public health and business impacts. The nation must make strategic investments to ensure energy reliability and foster the resilience of electric grids to weather hazards like extreme heat.
Advancing Solutions for Energy Systems and Grid Security
Investments in resilience pay dividends, with every federal dollar spent on resilience returning $6 in societal benefits. For example, the DOE Grid Resilience State and Tribal Formula Grants, established by the Bipartisan Infrastructure Law (BIL), have strengthened grid infrastructure, developed innovative technologies, and improved community resilience against extreme weather. It is essential that funds for this program, as well as other BIL and Inflation Reduction Act initiatives, continue to be disbursed.
To build heat resilience in communities across this nation, Congress must establish the HeatSmart Grids Initiative as a partnership between DOE, FEMA, HHS, the Federal Energy Regulatory Commission (FERC), NERC, and the Cybersecurity and Infrastructure Security Agency (CISA). This program should (i) perform national audits of energy security and building-stock preparedness for outages, (ii) map energy resilience assets such as long-term energy storage and microgrids, (iii) leverage technologies for minimizing grid loads such as smart grids and virtual power plants, and (iv) coordinate protocols with FEMA’s Community Lifelines and CISA’s Critical Infrastructure for emergency response. This initiative will ensure electric grids are prepared for extreme heat, including the risk of energy system failures during extreme heat and the necessary emergency and public health responses.
Challenge & Opportunity: Increasing Household and Business Energy Costs
As temperatures rise, so do household and business energy bills to cover cooling costs. This escalation can be particularly challenging for low-income individuals, schools, and small businesses operating on thin margins. For businesses, especially small enterprises, power outages, equipment failures, and interruptions in the supply chain become more frequent and severe due to extreme weather, negatively affecting production and distribution. One in six U.S. households (21.2 million people) find themselves behind on their energy bills, which increases the risk of utility shut-offs. One in five households report reducing or forgoing food and medicine to pay their energy bills. Families, school districts, and business owners need active and passive cooling approaches to meet demands without increasing costs.
Advancing Solutions for Businesses, Households, and Vital Facilities
Affordably cooled homes, businesses, and schools are crucial to sustaining our economy. To prepare the nation’s housing and infrastructure for rising temperatures, the federal government should:
- Expand the Weatherization Assistance Program (WAP), the Low-Income Home Energy Assistance Program, existing rebates and tax credits (including HER, HEAR, 25C, 179D, 45L, Direct Pay) to include passive cooling technology such as cool walls, pavements, and roofs (H.R. 9894). Revise 25C to be refundable at purchase to increase accessibility to low-income households.
- Authorize a Weatherization Readiness Program (H.R. 8721) to address structural, plumbing, roofing, and electrical issues and environmental hazards with dwelling units to make them eligible for WAP.
- Direct the DOE to work with its WAP contractors to ensure home energy audits consider passive cooling interventions, such as cool walls and roofs, strategically placing trees to provide shading and high-efficiency windows.
- Extend the National Initiative to Advance Building Codes (NIABC) to include (i) the development of codes and metrics for sustainable and passive cooling, shade, materials selection, and thermal comfort and (ii) the identification of opportunities to accelerate state and local adoption of code language for extreme heat adaptation. Partner with the National Institute of Standards and Technology to create an Extreme Heat clearinghouse for model building codes.
- Authorize an extension to the DOE Renew America’s Schools Program to continue funding cost-saving, energy-efficient upgrades in K-12 schools.
- Provide supportive appropriations to heat-critical programs at DOE, including: the Affordable Home Energy Shot, State and Community Energy Programs (SCEP), Office of Clean Energy Demonstrations (OCED), Office of Energy Efficiency & Renewable Energy (DOE), and the Home Energy Rebates program
The Federation of American Scientists: Who We Are
At the Federation of American Scientists (FAS), we envision a world where the federal government deploys cutting-edge science, technology, ideas, and talent to solve and address the impacts of extreme heat. We bring expertise in embedding science, data, and technology into government decision-making and a strong network of subject matter experts in extreme heat, both inside and outside of government. Through our 2025 Heat Policy Agenda and broader policy library, FAS is positioned to help ensure that public policy meets the challenges of living with extreme heat.
Consider FAS a resource for…
- Understanding evidence-based policy solutions
- Directing members and staff to relevant academic research
- Connecting with issue experts to develop solutions that can immediately address the impacts of extreme heat
We are tackling this crisis with initiative, creativity, experimentation, and innovation, serving as a resource on environmental health policy issues. Feel free to always reach out to us:
Emerging Technologies
Resilient Communities,
Extreme Weather,
Inclusive Innovation & Technology
Reforming the Federal Advisory Committee Landscape for Improved Evidence-based Decision Making and Increasing Public Trust
Federal Advisory Committees (FACs) are the single point of entry for the American public to provide consensus-based advice and recommendations to the federal government. These Advisory Committees are composed of experts from various fields who serve as Special Government Employees (SGEs), attending committee meetings, writing reports, and voting on potential government actions.
Advisory Committees are needed for the federal decision-making process because they provide additional expertise and in-depth knowledge for the Agency on complex topics, aid the government in gathering information from the public, and allow the public the opportunity to participate in meetings about the Agency’s activities. As currently organized, FACs are not equipped to provide the best evidence-based advice. This is because FACs do not meet transparency requirements set forth by GAO: making pertinent decisions during public meetings, reporting inaccurate cost data, providing official meeting documents publicly available online, and more. FACs have also experienced difficulty with recruiting and retaining top talent to assist with decision making. For these reasons, it is critical that FACs are reformed and equipped with the necessary tools to continue providing the government with the best evidence-based advice. Specifically, advice as it relates to issues such as 1) decreasing the burden of hiring special government employees 2) simplifying the financial disclosure process 3) increasing understanding of reporting requirements and conflict of interest processes 4) expanding training for Advisory Committee members 5) broadening the roles of Committee chairs and designated federal officials 6) increasing public awareness of Advisory Committee roles 7) engaging the public outside of official meetings 8) standardizing representation from Committee representatives 9) ensuring that Advisory Committees are meeting per their charters and 10) bolstering Agency budgets for critical Advisory Committee issues.
Challenge and Opportunity
Protecting the health and safety of the American public and ensuring that the public has the opportunity to participate in the federal decision-making process is crucial. We must evaluate the operations and activities of federal agencies that require the government to solicit evidence-based advice and feedback from various experts through the use of federal Advisory Committees (FACs). These Committees are instrumental in facilitating transparent and collaborative deliberation between the federal government, the advisory body, and the American public and cannot be done through the use of any other mechanism. Advisory Committee recommendations are integral to strengthening public trust and reinforcing the credibility of federal agencies. Nonetheless, public trust in government has been waning and efforts should be made to increase public trust. Public trust is known as the pillar of democracy and fosters trust between parties, particularly when one party is external to the federal government. Therefore, the use of Advisory Committees, when appropriately used, can assist with increasing public trust and ensuring compliance with the law.
There have also been many success stories demonstrating the benefits of Advisory Committees. When Advisory Committees are appropriately staffed based on their charge, they can decrease the workload of federal employees, assist with developing policies for some of our most challenging issues, involve the public in the decision-making process, and more. However, the state of Advisory Committees and the need for reform have been under question, and even more so as we transition to a new administration. Advisory Committees have contributed to the improvement in the quality of life for some Americans through scientific advice, as well as the monitoring of cybersecurity. For example, an FDA Advisory Committee reviewed data and saw promising results for the treatment of sickle cell disease (SCD) which has been a debilitating disease with limited treatment for years. The Committee voted in favor of gene therapy drugs Casgevy and Lyfgenia which were the first to be approved by the FDA for SCD.
Under the first Trump administration, Executive Order (EO) 13875 resulted in a significant decrease in the number of federal advisory meetings. This limited agencies’ ability to convene external advisors. Federal science advisory committees met less during this administration than any prior administration, met less than what was required from their charter, disbanded long standing Advisory Committees, and scientists receiving agency grants were barred from serving on Advisory Committees. Federal Advisory Committee membership also decreased by 14%, demonstrating the issue of recruiting and retaining top talent. The disbandment of Advisory Committees, exclusion of key scientific external experts from Advisory Committees, and burdensome procedures can potentially trigger severe consequences that affect the health and safety of Americans.
Going into a second Trump administration, it is imperative that Advisory Committees have the opportunity to assist federal agencies with the evidence-based advice needed to make critical decisions that affect the American public. The suggested reforms that follow can work to improve the overall operations of Advisory Committees while still providing the government with necessary evidence-based advice. With successful implementation of the following recommendations, the federal government will be able to reduce administrative burden on staff through the recruitment, onboarding, and conflict of interest processes.
The U.S. Open Government Initiative encourages the promotion and participation of public and community engagement in governmental affairs. However, individual Agencies can and should do more to engage the public. This policy memo identifies several areas of potential reform for Advisory Committees and aims to provide recommendations for improving the overall process without compromising Agency or Advisory Committee membership integrity.
Plan of Action
The proposed plan of action identifies several policy recommendations to reform the federal Advisory Committee (Advisory Committee) process, improving both operations and efficiency. Successful implementation of these policies will 1) improve the Advisory Committee member experience, 2) increase transparency in federal government decision-making, and 3) bolster trust between the federal government, its Advisory Committees, and the public.
Streamline Joining Advisory Committees
Recommendation 1. Decrease the burden of hiring special government employees in an effort to (1) reduce the administrative burden for the Agency and (2) encourage Advisory Committee members, who are also known as special government employees (SGEs), to continue providing the best evidence-based advice to the federal government through reduced onerous procedures
The Ethics in Government Act of 1978 and Executive Order 12674 lists OGE-450 reporting as the required public financial disclosure for all executive branch and special government employees. This Act provides the Office of Government Ethics (OGE) the authority to implement and regulate a financial disclosure system for executive branch and special government employees whose duties have “heightened risk of potential or actual conflicts of interest”. Nonetheless, the reporting process becomes onerous when Advisory Committee members have to complete the OGE-450 before every meeting even if their information remains unchanged. This presents a challenge for Advisory Committee members who wish to continue serving, but are burdened by time constraints. The process also burdens federal staff who manage the financial disclosure system.
Policy Pathway 1. Increase funding for enhanced federal staffing capacity to undertake excessive administrative duties for financial reporting.
Policy Pathway 2. All federal agencies that deploy Advisory Committees can conduct a review of the current OGC-450 process, budget support for this process, and work to develop an electronic process that will eliminate the use of forms and allow participants to select dropdown options indicating if their financial interests have changed.
Recommendation 2. Create and use public platforms such as OpenPayments by CMS to (1) aid in simplifying the financial disclosure reporting process and (2) increase transparency for disclosure procedures
Federal agencies should create a financial disclosure platform that streamlines the process and allows Advisory Committee members to submit their disclosures and easily make updates. This system should also be created to monitor and compare financial conflicts. In addition, agencies that utilize the expertise of Advisory Committees for drugs and devices should identify additional ways in which they can promote financial transparency. These agencies can use Open Payments, a system operated by Centers for Medicare & Medicaid Services (CMS), to “promote a more financially transparent and accountable healthcare system”. The Open Payments system makes payments from medical and drug device companies to individuals, healthcare providers, and teaching hospitals accessible to the public. If for any reason financial disclosure forms are called into question, the Open Payments platform can act as a check and balance in identifying any potential financial interests of Advisory Committee members. Further steps that can be taken to simplify the financial disclosure process would be to utilize conflict of interest software such as Ethico which is a comprehensive tool that allows for customizable disclosure forms, disclosure analytics for comparisons, and process automation.
Policy Pathway. The Office of Government Ethics should require all federal agencies that operate Advisory Committees to develop their own financial disclosure system and include a second step in the financial disclosure reporting process as due diligence, which includes reviewing the Open Payments by CMS system for potential financial conflicts or deploying conflict of interest monitoring software to streamline the process.
Streamline Participation in an Advisory Committee
Recommendation 3. Increase understanding of annual reporting requirements for conflict of interest (COI)
Agencies should develop guidance that explicitly states the roles of Ethics Officers, also known as Designated Agency Ethics Officials (DAEO), within the federal government. Understanding the roles and responsibilities of Advisory Committee members and the public will help reduce the spread of misinformation regarding the purpose of Advisory Committees. In addition, agencies should be encouraged by the Office of Government Ethics to develop guidance that indicates the criteria for inclusion or exclusion of participation in Committee meetings. Currently, there is no public guidance that states what types of conflicts of interests are granted waivers for participation. Full disclosure of selection and approval criteria will improve transparency with the public and draw clear delineations between how Agencies determine who is eligible to participate.
Policy Pathway. Develop conflict of interest (COI) and financial disclosure guidance specifically for SGEs that states under what circumstances SGEs are allowed to receive waivers for participation in Advisory Committee meetings.
Recommendation 4. Expand training for Advisory Committee members to include (1) ethics and (2) criteria for making good recommendations to policymakers
Training should be expanded for all federal Advisory Committee members to include ethics training which details the role of Designated Agency Ethics Officials, rules and regulations for financial interest disclosures, and criteria for making evidence-based recommendations to policymakers. Training for incoming Advisory Committee members ensures that all members have the same knowledge base and can effectively contribute to the evidence-based recommendations process.
Policy Pathway. Agencies should collaborate with the OGE and Agency Heads to develop comprehensive training programs for all incoming Advisory Committee members to ensure an understanding of ethics as contributing members, best practices for providing evidence-based recommendations, and other pertinent areas that are deemed essential to the Advisory Committee process.
Leverage Advisory Committee Membership
Recommendation 5. Uplifting roles of the Committee Chairs and Designated Federal Officials
Expanding the roles of Committee Chairs and Designated Federal Officers (DFOs) may assist federal Agencies with recruiting and retaining top talent and maximizing the Committee’s ability to stay abreast of critical public concerns. Considering the fact that the General Services Administration has to be consulted for the formation of new Committees, renewal, or alteration of Committees, they can be instrumental in this change.
Policy Pathway. The General Services Administration (GSA) should encourage federal Agencies to collaborate with Committee Chairs and DFOs to recruit permanent and ad hoc Committee members who may have broad network reach and community ties that will bolster trust amongst Committees and the public.
Recommendation 6. Clarify intended roles for Advisory Committee members and the public
There are misconceptions among the public and Advisory Committee members about Advisory Committee roles and responsibilities. There is also ambiguity regarding the types of Advisory Committee roles such as ad hoc members, consulting, providing feedback for policies, or making recommendations.
Policy Pathway. GSA should encourage federal Agencies to develop guidance that delineates the differences between permanent and temporary Advisory Committee members, as well as their roles and responsibilities depending on if they’re providing feedback for policies or providing recommendations for policy decision-making.
Recommendation 7. Utilize and engage expertise and the public outside of public meetings
In an effort to continue receiving the best evidence-based advice, federal Agencies should develop alternate ways to receive advice outside of public Committee meetings. Allowing additional opportunities for engagement and feedback from Committee experts or the public will allow Agencies to expand their knowledge base and gather information from communities who their decisions will affect.
Policy Pathway. The General Services Administration should encourage federal Agencies to create opportunities outside of scheduled Advisory Committee meetings to engage Committee members and the public on areas of concern and interest as one form of engagement.
Recommendation 8. Standardize representation from Committee representatives (i.e., industry), as well as representation limits
The Federal Advisory Committee Act (FACA) does not specify the types of expertise that should be represented on all federal Advisory Committees, but allows for many types of expertise. Incorporating various sets of expertise that are representative of the American public will ensure the government is receiving the most accurate, innovative, and evidence-based recommendations for issues and products that affect Americans.
Policy Pathway. Congress should include standardized language in the FACA that states all federal Advisory Committees should include various sets of expertise depending on their charge. This change should then be enforced by the GSA.
Support a Vibrant and Functioning Advisory Committee System
Recommendation 9. Decrease the burden to creating an Advisory Committee and make sure Advisory Committees are meeting per their charters
The process to establish an Advisory Committee should be simplified in an effort to curtail the amount of onerous processes that lead to a delay in the government receiving evidence based advice.
Advisory Committee charters state the purpose of Advisory Committees, their duties, and all aspirational aspects. These charters are developed by agency staff or DFOs with consultation from their agency Committee Management Office. Charters are needed to forge the path for all FACs.
Policy Pathway. Designated Federal Officers (DFOs) within federal agencies should work with their Agency head to review and modify steps to establishing FACs. Eliminate the requirement for FACs to require consultation and/or approval from GSA for the formation, renewal, or alteration of Advisory Committees.
Recommendation 10. Bolster agency budgets to support FACs on critical issues where regular engagement and trust building with the public is essential for good policy
Federal Advisory Committees are an essential component to receive evidence-based recommendations that will help guide decisions at all stages of the policy process. These Advisory Committees are oftentimes the single entry point external experts and the public have to comment and participate in the decision-making process. However, FACs take considerable resources to operate depending on the frequency of meetings, the number of Advisory Committee members, and supporting FDA staff. Without proper appropriations, they have a diminished ability to recruit and retain top talent for Advisory Committees. The Government Accountability Office (GAO) reported that in 2019, approximately $373 million dollars was spent to operate a total of 960 federal Advisory Committees. Some Agencies have experienced a decrease in the number of Advisory Committee convenings. Individual Agency heads should conduct a budget review of average operating and projected costs and develop proposals for increased funding to submit to the Appropriations Committee.
Policy Pathway. Congress should consider increasing appropriations to support FACs so they can continue to enhance federal decision-making, improve public policy, boost public credibility, and Agency morale.
Conclusion
Advisory Committees are necessary to the federal evidence-based decision-making ecosystem. Enlisting the advice and recommendations of experts, while also including input from the American public, allows the government to continue making decisions that will truly benefit its constituents. Nonetheless, there are areas of FACs that can be improved to ensure it continues to be a participatory, evidence-based process. Additional funding is needed to compensate the appropriate Agency staff for Committee support, provide potential incentives for experts who are volunteering their time, and finance other expenditures.
With reform of Advisory Committees, the process for receiving evidence-based advice will be streamlined, allowing the government to receive this advice in a faster and less burdensome manner. Reform will be implemented by reducing the administrative burden for federal employees through the streamlining of recruitment, financial disclosure, and reporting processes.
Protecting Infant Nutrition Security:
Shifting the Paradigm on Breastfeeding to Build a Healthier Future for all Americans
The health and wellbeing of American babies have been put at risk in recent years, and we can do better. Recent events have revealed deep vulnerabilities in our nation’s infant nutritional security. For example: Pandemic-induced disruptions in maternity care practices that support the establishment of breastfeeding; the infant formula recall and resulting shortage; and a spate of weather-related natural disasters have demonstrated infrastructure gaps and a lack of resilience to safety and supply chain challenges. All put babies in danger during times of crisis.
Breastfeeding is foundational to lifelong health and wellness, but systemic barriers prevent many families from meeting their breastfeeding goals. The policies and infrastructure surrounding postpartum families often limit their ability to succeed in breastfeeding. Despite important benefits, new data from the CDC shows that while 84.1% of infants start out breastfeeding, these numbers fall dramatically in the weeks after birth, with only 57.5% of infants breastfeeding exclusively at one month of age. Disparities persist across geographic location, and other sociodemographic factors, including race/ethnicity, maternal age, and education. Breastfeeding rates in North America are the lowest in the world. Longstanding evidence shows that it is not a lack of desire but rather a lack of support, access, and resources that creates these barriers.
This administration has an opportunity to take a systems approach to increasing support for breastfeeding and making parenting easier for new mothers. Key policy changes to address systemic barriers include providing guidance to states on expanding Medicaid coverage of donor milk, building breastfeeding support and protection into the existing emergency response framework at the Federal Emergency Management Agency, and expressing support for establishing a national paid leave program.
Policymakers on both sides of the aisle agree that no baby should ever go hungry, as evidenced by the bipartisan passage of recent breastfeeding legislation (detailed below) and widely supported regulations. However, significant barriers remain. This administration has the power to address long-standing inequities and set the stage for the next generation of parents and infants to thrive. Ensuring that every family has the support they need to make the best decisions for their child’s health and wellness benefits the individual, the family, the community, and the economy.
Challenge and Opportunity
Breastfeeding plays an essential role in establishing good nutrition and healthy weight, reducing the risk of chronic disease and infant mortality, and improving maternal and infant health outcomes. Breastfed children have a decreased risk of obesity, type 1 and 2 diabetes, asthma, and childhood leukemia. Women who breastfeed reduce their risk of specific chronic diseases, including type 2 diabetes, cardiovascular disease, and breast and ovarian cancers. On a relational level, the hormones produced while breastfeeding, like oxytocin, enhance the maternal-infant bond and emotional well-being. The American Academy of Pediatrics recommends infants be exclusively breastfed for approximately six months with continued breastfeeding while introducing complementary foods for two years or as long as mutually desired by the mother and child.
Despite the well-documented health benefits of breastfeeding, deep inequities in healthcare, community, and employment settings impede success. Systemic barriers disproportionately impact Black, Indigenous, and other communities of color, as well as families in rural and economically distressed areas. These populations already bear the weight of numerous health inequities, including limited access to nutritious foods and higher rates of chronic disease—issues that breastfeeding could help mitigate.
Breastfeeding Saves Dollars and Makes Sense
Low breastfeeding rates in the United States cost our nation millions of dollars through higher health system costs, lost productivity, and higher household expenditures. Not breastfeeding is associated with economic losses of about $302 billion annually or 0.49% of world gross national income. At the national level, improving breastfeeding practices through programs and policies is one of the best investments a country can make, as every dollar invested is estimated to result in a $35 economic return.
In the United States, chronic disease management results in trillions of dollars in annual healthcare costs, which increased breastfeeding rates could help reduce. In the workplace setting, employers see significant cost savings when their workers are able to maintain breastfeeding after returning to work. Increased breastfeeding rates are also associated with reduced environmental impact and associated expenses. Savings can be seen at home as well, as following optimal breastfeeding practices reduces household expenditures. Investments in infant nutrition last a lifetime, paying long-term dividends critical for economic and human development. Economists have completed cost-benefit analyses, finding that investments in nutrition are one of the best value-for-money development actions, laying the groundwork for the success of investments in other sectors.
{kind=link}
Ongoing trends in breastfeeding outcomes indicate that there are entrenched policy-level challenges and barriers that need to be addressed to ensure that all infants have an opportunity to benefit from access to human milk. Currently, for too many families, the odds are stacked against them. It’s not a question of individual choice but one of systemic injustice. Families are often forced into feeding decisions that do not reflect their true desires due to a lack of accessible resources, support, and infrastructure.
While the current landscape is rife with challenges, the solutions are known and the potential benefits are tremendous. This administration has the opportunity to realize these benefits and implement a smart and strategic response to the urgent situation that our nation is facing just as the political will is at an all-time high.
The History of Breastfeeding Policy
In the late 1960s and early 1970s less than 30 percent of infants were breastfed. The concerted efforts of individuals and organizations and the emergence of the field of lactation have worked to counteract or remove many barriers, and policymakers have sent a clear and consistent message that breastfeeding is bipartisan. This is evident in the range of recent lactation-friendly legislation, including:
- Bottles and Breastfeeding Equipment Screening Act (BABES Act) in 2016
- Friendly Airports for Mothers (FAM) Act in 2018
- Fairness for Breastfeeding Mothers Act in 2019
- Providing Urgent Maternal Protections (PUMP) for Nursing Mothers Act in 2022
Administrative efforts ranging from the Business Case for Breastfeeding to The Surgeon General’s Call to Action to Support Breastfeeding and the armed services updates on uniform requirements for lactating soldiers demonstrate a clear commitment to breastfeeding support across the decades.
These policy changes have made a difference. But additional attention and investment, with a particular focus on the birth and early postpartum period, as well as during and after emergencies, is needed to secure the potential health and economic benefits of comprehensive societal support for breastfeeding. This administration can take considerable steps toward improving U.S. health and wellness and protecting infant nutrition security.
Plan of Action
A range of federal agencies coordinate programs, services, and initiatives impacting the breastfeeding journey for new parents. Expanding and building on existing efforts through the following steps can help address some of today’s most pressing barriers to breastfeeding.
Each of the recommended actions can be implemented independently and would create meaningful, incremental change for families. However, a comprehensive approach that implements all these recommendations would create the marked shift in the landscape needed to improve breastfeeding initiation and duration rates and establish this administration as a champion for breastfeeding families.
Recommendation 1. Increase access to pasteurized donor human milk by directing the Centers for Medicare & Medicaid Services (CMS) to provide guidance to states on expanding Medicaid coverage.
Pasteurized donor human milk is lifesaving for vulnerable infants, particularly those born preterm or with serious health complications. Across the United States, milk banks gently pasteurize donated human milk and distribute it to fragile infants in need. This lifesaving liquid gold reduces mortality rates, lowers healthcare costs, and shortens hospital stays. Specifically, the use of donor milk is associated with increased survival rates and lowered rates of infections, sepsis, serious lung disease, and gastrointestinal complications. In 2022, there were 380,548 preterm births in the United States, representing 10.4% of live births, so the potential for health and cost savings is substantial. Data from one study shows that the cost of a neonatal intensive care unit stay for infants at very low birth weight is nearly $220,000 for 56 days. The use of donor human milk can reduce hospital length of stay by 18-50 days by preventing the development of necrotizing enterocolitis in preterm infants. The benefits of human milk extend beyond the inpatient stay, with infants receiving all human milk diets in the NICU experiencing fewer hospital readmissions and better overall long-term outcomes.
Although donor milk has important health implications for vulnerable infants in all communities and can result in significant economic benefit, donor milk is not equitably accessible. While milk banks serve all states, not all communities have easy access to donated human milk. Moreover, many insurers are not required to cover the cost, creating significant barriers to access and contributing to racial and geographic disparities.
To ensure that more babies in need have access to lifesaving donor milk, the administration should work with CMS to expand donor milk coverage under state Medicaid programs. Medicaid covers approximately 40% of all US births and 50% of all early preterm births. Medicaid programs in at least 17 states and the District of Columbia already include coverage of donor milk. The administration can expand access to this precious milk, help reduce health care costs, and address racial and geographic disparities by releasing guidance for the remaining states regarding coverage options in Medicaid.
Recommendation 2. Include infant feeding in Federal Emergency Management Agency (FEMA) emergency planning and response.
Infants and children are among the most vulnerable in an emergency, so it is critical that their unique needs are considered and included in emergency planning and response guidance. Breastfeeding provides clean, optimal nutrition, requires no fuel, water, or electricity, and is available, even in the direst circumstances. Human milk contains antibodies that fight infection, including diarrhea and respiratory infections common among infants in emergency situations. Yet efforts to protect infant and young child feeding in emergencies are sorely lacking, particularly in the immediate aftermath of disasters and emergencies.
Ensuring access to lactation support and supplies as part of emergency response efforts is essential for protecting the health and safety of infants. Active support and coordination between federal, state, and local governments, the commercial milk formula industry, lactation support providers, and all other relevant actors involved in response to emergencies is needed to ensure safe infant and young child feeding practices and equitable access to support. There are two simple, cost-effective steps that FEMA can take to protect breastfeeding, preserve resources, and thus save additional lives during emergencies.
- Require FEMA to participate in the Federal Interagency Breastfeeding Workgroup, a collection of federal agencies that come together to connect and collaborate on breastfeeding issues, to ensure coordination across agencies.
- Update the FEMA Public Assistance Program and Policy Guide (PAPPG) to include breastfeeding and lactation as a functional need in the section on Congregate Shelter Services so that emergency response efforts can include services from lactation support providers and be reimbursed for the associated costs.
Recommendation 3. Expand access to paid family & medical leave by including paid leave as a priority in the President’s Budget and supporting the efforts of the bipartisan, bicameral congressional Paid Leave Working Group.
Employment policies in the United States make breastfeeding harder than it needs to be. The United States is one of the only countries in the world without a national paid family and medical leave program. Many parents return to work quickly after birth, before a strong breastfeeding relationship is established, because they cannot afford to take unpaid leave or because they do not qualify for paid leave programs with their employer or through state or local programs. Nearly 1 in 4 employed mothers return to work within two weeks of childbirth.
Paid family leave programs make it possible for employees to take time for childbirth recovery, bond with their baby, establish feeding routines, and adjust to life with a new child without threatening their family’s economic well-being. This precious time provides the foundation for success, contributing to improved rates of breastfeeding initiation and duration, yet only a small portion of workers are able to access it. There are significant disparities in access to paid leave among racial and ethnic groups, with Black and Hispanic employees less likely than their white non-Hispanic counterparts to have access to paid parental leave. There are similar disparities in breastfeeding outcomes among racial groups.
The momentum is building substantially to improve the paid family and medical leave landscape in the United States. Thirteen states and the District of Columbia have established mandatory state paid family leave systems. Supporting paid leave has become an important component of candidate campaign plans, and bipartisan support for establishing a national program remains strong among voters. The formation of Bipartisan Paid Family Leave Working Groups in both the House and Senate demonstrate commitment from policymakers on both sides of the aisle.
By directing the Office of Management and Budget to include funding for paid leave in the President’s Budget recommendation and working collaboratively with the Congressional Paid Leave Working Groups, the administration can advance federal efforts to increase access to paid family and medical leave, improving public health and helping American businesses.
Conclusion
These three strategies offer the opportunity for the White House to make an immediate and lasting impact by protecting infant nutrition security and addressing disparities in breastfeeding rates, on day one of the Presidential term. A systems approach that utilizes multiple strategies for integrating breastfeeding into existing programs and efforts would help shift the paradigm for new families by addressing long-standing barriers that disproportionately affect marginalized communities—particularly Black, Indigenous, and families of color. A clear and concerted effort from the Administration, as outlined, offers the opportunity to benefit all families and future generations of American babies.
The administration’s focused and strategic efforts will create a healthier, more supportive world for babies, families, and breastfeeding parents, improve maternal and child health outcomes, and strengthen the economy. This administration has the chance to positively shape the future for generations of American families, ensuring that every baby gets the best possible start in life and that every parent feels empowered and supported.
Now is the time to build on recent momentum and create a world where families have true autonomy in infant feeding decisions. A world where paid family leave allows parents the time to heal, bond, and establish feeding routines; communities provide equitable access to donor milk; and federal, state, and local agencies have formal plans to protect infant feeding during emergencies, ensuring no baby is left vulnerable. Every family deserves to feel empowered and supported in making the best choices for their children, with equitable access to resources and support systems.
This policy memo was written with support from Suzan Ajlouni, Public Health Writing Specialist at the U.S. Breastfeeding Committee. The policy recommendations have been identified through the collective learning, idea sharing, and expertise of USBC members and partners.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
Rather than being a matter of personal choice, infant feeding practice is informed by circumstance and level (or lack) of support. When roadblocks exist at every turn, families are backed into a decision because the alternatives are not available, attainable, or viable. United States policies and infrastructure were not built with the realities of breastfeeding in mind. Change is needed to ensure that all who choose to breastfeed are able to meet their personal breastfeeding goals, and society at large reaps the beneficial social and economic outcomes.
The Fiscal Year 2024 President’s Budget proposed to establish a national, comprehensive paid family and medical leave program, providing up to 12 weeks of leave to allow eligible workers to take time off to care for and bond with a new child; care for a seriously ill loved one; heal from their own serious illness; address circumstances arising from a loved one’s military deployment; or find safety from domestic violence, sexual assault, or stalking. The budget recommendation included $325 billion for this program. It’s important to look at this with the return on investment in mind, including improved labor force attachment and increased earnings for women; better outcomes and reduced health care costs for ill, injured, or disabled loved ones; savings to other tax-funded programs, including Medicaid, SNAP, and other forms of public assistance; and national economic growth, jobs growth, and increased economic activity.
There are a variety of national monitoring and surveillance efforts tracking breastfeeding initiation, duration, and exclusivity rates that will inform how well these actions are working for the American people, including the National Immunization Survey (NIS), Pregnancy Risk Assessment and Monitoring System (PRAMS), Infant Feeding Practices Study, and National Vital Statistics System. The CDC Breastfeeding Report card is published biannually to bring these key data points together and place them into context. Significant improvements in the data have already been seen across recent decades, with breastfeeding initiation rates increasing from 73.1 percent in 2004 to 84.1 percent in 2021.
The U.S. Breastfeeding Committee is a coalition bringing together approximately 140 organizations from coast to coast representing the grassroots to the treetops – including federal agencies, national, state, tribal, and territorial organizations, and for-profit businesses – that support the USBC mission to create a landscape of breastfeeding support across the United States. Nationwide, a network of hundreds of thousands of grassroots advocates from across the political spectrum support efforts like these. Together, we are committed to ensuring that all families in the U.S. have the support, resources, and accommodations to achieve their breastfeeding goals in the communities where they live, learn, work, and play. The U.S. Breastfeeding Committee and our network stand ready to work with the administration to advance this plan of action.
Supporting Device Reprocessing to Reduce Waste in Health Care
The U.S. healthcare system produces 5 million tons of waste annually, or approximately 29 pounds per hospital bed daily. Roughly 80 percent of the healthcare industry’s carbon footprint comes from the production, transportation, use, and disposal of single-use devices (SUDs), which are pervasive in the hospital. Notably, 95% of the environmental impact of single-use medical products results from the production of those products.
While the Food and Drug Administration (FDA) oversees new devices being brought to market, it is up to the manufacturer to determine whether a device will be marketed as single-use or multiple-use. Manufacturers have a financial incentive to market devices for “single-use” or “disposable” as marketing a device as reusable requires expensive cleaning validations.
In order to decrease healthcare waste and environmental impact, FDA leads on identifying reusable devices that can be safely reprocessed and incentivizing manufacturers to test the reprocessing of their device. This will require the FDA to strengthen its management of single-use and reusable device labeling. Further, the Veterans Health Administration, the nation’s largest healthcare system, should reverse the prohibition on reprocessed SUDs and become a national leader in the reprocessing of medical devices.
Challenge and Opportunity
While healthcare institutions are embracing decarbonization and waste reduction plans, they cannot do this effectively without addressing the enormous impact of single-use devices (SUDs). The majority of research literature concludes that SUDs are associated with higher levels of environmental impact than reusable products.
FDA regulations governing SUD reprocessing make it extremely challenging for hospitals to reprocess low-risk SUDs, which is inconsistent with the FDA’s “least burdensome provisions.” The FDA requires hospitals or commercial SUD reprocessing facilities to act as the device’s manufacturer, meaning they must follow the FDA’s rules for medical device manufacturers’ requirements and take on the associated liabilities. Hospitals are not keen to take on the liability of a manufacturer, yet commercial reprocessors do not offer many lower-risk devices that can be reprocessed.
As a result, hospitals and clinics are no longer willing to sterilize SUDs through methods like autoclaving even despite documentation showing that sterilization is safe and precedent showing that similar devices have been safely sterilized and reused for many years without adverse events. Many devices, including pessaries for pelvic organ prolapse and titanium phacoemulsification tips for cataract surgery, can be safely reprocessed in their clinical use. These products, given their risk profile, need not be subject to the FDA’s full medical device manufacturer requirements.
Further, manufacturers are incentivized to bring SUDs to market quicker than those that may be reprocessed. Manufacturers often market devices as single-use solely because the original manufacturer chose not to conduct expensive cleaning and sterilization validations, not because such cleaning and sterilization validations cannot be done. FDA regulations that govern SUDs should be better tailored to each device so that clinicians on the frontlines can provide appropriate and environmentally sustainable health care.
Reprocessed devices cost 25 to 40% less. Thus, the use of reprocessed SUDs can reduce costs in hospitals significantly — about $465 million in 2023. Per the Association of Medical Device Reprocessors, if the reprocessing practices of the top 10% performing hospitals were maximized across all hospitals that use reprocessed devices, U.S. hospitals could have saved an additional $2.28 billion that same year. Indeed, enabling and encouraging the use of reprocessed SUDs can also yield significant cost reductions without compromising patient care.
Plan of Action
As the FDA began regulating SUD reprocessing in 2000, it is imperative that the FDA take the lead on creating a clear, streamlined process for clearing or approving reusable devices in order to ensure the safety and efficacy of reprocessed devices. These recommendations would permit healthcare systems to reprocess and reuse medical devices without fear of noncompliance by the Joint Commission or Centers for Medicare and Medicaid Services that reply on FDA regulations. Further, the nation’s largest healthcare system, the Veterans Health Administration, should become a leader in medical device reprocessing, and lead on showcasing the standard of practice for sustainable healthcare.
- FDA should publish a list of SUDs that have a proven track record of safe reprocessing to empower hospitals to reduce waste, costs, and environmental impact without compromising patient safety. The FDA should change the labels of single-use devices to multi-use when reuse by hospitals is possible and validated via clinical studies, as the “single-use” label has promoted the mistaken belief that SUDs cannot be safely reprocessed. Per the FDA, the single-use label simply means a given device has not undergone the original equipment manufacturer (OEM) validation tests necessary to label a device “reusable.” The label does not mean the device cannot be cleared for reprocessing.
- In order to help governments and healthcare systems prioritize the environmental and cost benefits of reusable devices over SUDs, FDA should incentivize applications of reusable or commercially reprocessable devices, such as through expediting review. The FDA can also incentivize use of reprocessed devices through payments to hospitals for meeting reprocessing benchmarks.
- The FDA should not subject low-risk devices that can be safely reprocessed for clinical use to full device manufacturer requirements. The FDA should further encourage healthcare procurement staff by creating an accessible database of devices cleared for reprocessing and alerting healthcare systems about regulated reprocessing options. In doing so, the FDA can help reduce the burden on hospitals in reprocessing low-risk SUDs and encourage healthcare systems to sterilize SUDs through methods like autoclaving.
- As the only major health system in the U.S. to prohibit the use of reprocessed SUDs, the U.S. Veterans Health Administration should reverse its prohibition as soon as possible. This prohibition likely remains because of outdated determinations of risks, which comes at major costs for the environment and Americans. Doing so would be consistent with the FDA’s conclusions that reprocessed SUDs are safe and effective.
- FDA should recommend that manufacturers publicly report the materials used in the composition of devices so that end-users can more easily compare products and determine the environmental impact of devices. As explained by AMDR, some Original Equipment Manufacturer (OEM) practices discourage or fully prevent the use of reprocessed devices. It is imperative that the FDA vigorously track and impede these practices. Not only will requiring public reporting device composition help healthcare buyers make more informed decisions, it will also help promote a more circular economy that uplifts sustainability efforts.
Conclusion
To decrease costs, waste, and environmental impact, the healthcare sector urgently needs to increase its use of reusable devices. One of the largest barriers is FDA requirements that result in needlessly stringent requirements of hospitals, hindering the adoption of less wasteful, less costly reprocessed devices.
FDA’s critical role in medical device labeling, clearing, or approving more devices as reusable, has down market implications and influences many other regulatory and oversight bodies, including the Centers for Medicare & Medicaid Services (CMS), the Association for the Advancement of Medical Instrumentation (AAMI), the Joint Commission, hospitals, health care offices, and health care providers. It is essential for the FDA to step up and take the lead in revising the device reprocessing pipeline.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
A Peer Support Service Integrated Into the 988 Lifeline
A peer support option should be integrated into the 988 Suicide and Crisis Lifeline so that 988 service users can choose to connect with specialists based on a shared lived experience. As people and communities become more siloed, the risk of mortality and morbidity increases. Social connectedness is a critical protective factor. A peer support service would allow individuals to receive support built on a lived experience that is common to both the service user and the specialist. It should be free and easily accessible through phone call and text messaging. This service is especially timely, following the 2020 rollout of the 988 Suicide and Crisis Lifeline, as well as recent peer support initiatives at the federal and state levels.
While the efficacy of peer support is most known with mental illness, it has successfully helped a range of individuals including cancer patients, people experiencing homelessness, racial minorities, veterans, and formerly incarcerated people. The peer support service should provide support for all kinds of lived experience, including experiences with: disability resulting from poor physical or mental health, substance use, suicidal ideation, veteran-connected disability, financial insecurity, homelessness, domestic violence and family court, nonnuclear family structures or living alone, former incarceration, and belonging to racial or ethnic minority groups. The service should be a preventative intervention as well as complimentary assistance for those in recovery or treatment.
Challenge and Opportunity
The United States faces an “epidemic of loneliness and isolation.” While individuals from all backgrounds are afflicted, the most vulnerable and underserved members of society have suffered the most. A range of challenging circumstances cause this suffering, including poor physical or mental health, nontraditional living conditions, and historic inequality. Not having anyone who shares one’s lived experience is isolating. These challenging life circumstances take an emotional toll, and they become risk factors for a slew of physical and mental health conditions that increase morbidity and decrease life expectancy.
In addition to the social costs, these outcomes have an increasingly devastating economic impact. Health care costs are projected to account for 20% of the U.S. economy by 2031. Psychiatric hospitalizations are steadily increasing, and a shortage of psychiatric inpatient beds is rampant.
Social connectedness alleviates physical and mental burdens. Peer support offers a cost-effective intervention for prevention and recovery. It reduces spending on both physical and mental illnesses, and it reduces psychiatric hospitalizations, saving an average of $4.76 for every $1 spent on peer support. In a New York City-based peer support program, service users saved about $2,000 per month in Medicaid costs and had 2.9 fewer hospitalizations per year.
Telephone-based peer support has life-saving outcomes, too. Over telephone, peer support led to a 15% increase in women’s mammography screening rates, with the highest increase among women of low-to-middle income. Telephone-based peer support increased breastfeeding rates by 14% and reduced breastfeeding dissatisfaction by 10% among first-time mothers, and it led to a 10% change in diet among patients with heart disease. While peer support would not solve national crises of homelessness or rising healthcare costs, it would ease them by fostering community empowerment and self-reliance and reducing federal intervention.
In 2023, SAMHSA rolled out “National Model Standards for Peer Support Certification”. This guide provides recommendations for how each state can integrate its own “peer mental health workforce across all elements of the healthcare system.” In its current form, SAMHSA’s strategy targets lived experiences with substance use and mental health. A broader scope would assist and empower more underserved members of the community. Following the momentum of SAMHSA’s initiative, now is the most optimal time to integrate a peer support service into the 988 Lifeline.
Peer support exists in the U.S., but services are spread thin across private and nonprofit sectors. The Restoring Hope for Mental Health and Well-Being Act of 2022 authorized $1.7 trillion in funding until FY2027 for various health initiatives, including peer support mental health services. This funding relies on organizations and institutions to independently implement peer support services based on community needs. However, a fragmented system can result in underuse, limited accessibility, and a varied quality of service, with pockets of the United States lacking any service at all. It also poses privacy concerns about how individuals’ data are stored and used, as well as cybersecurity vulnerabilities with smaller organizations that may lack a robust security infrastructure.
A peer support service that is integrated into the 988 Lifeline would ensure that all Americans have equal access to a high-quality, confidential peer support network. The standardization of the 988 Lifeline is a prime example of successful implementation. Its transition from a 10-digit number to a three-digit dial led to a 33% average increase in in-state call volume over four months. Standardization shifted funding from primarily private and nonprofit initiatives and donations to stable public sector support. As a result, call pickup rates rose from 70% to 93%, and wait times dropped from 2 minutes 20 seconds to just 35 seconds.
The 988 Lifeline also adheres to privacy and confidentiality protocols in line with the Health Insurance Portability and Accountability Act (HIPAA) Security Rule. Under these protocols, the 988 Lifeline retains minimal information about callers and texters, and this information stays private and securely stored. A peer support service with similar safeguards would help both service users and peer support workers (PSWs) feel safe when sharing sensitive information.
Peer support for mental health has gained traction recently. The National Alliance for Mental Illness (NAMI) offers peer-to-peer courses, although these are currently limited in their duration and available locations. Last year, Congress expressed an interest in peer support mental health services: a “Supporting All Students Act” was proposed in the Senate for “peer and school-based mental health support.” This endeavor especially targets the escalating suicide crisis among youths in the form of a peer-to-peer suicide prevention. The Wisconsin Department of Public Instruction also recently introduced Peer-to-Peer Suicide Prevention Grants. The federal and state-level governments clearly recognize the value of peer support work. Still, they underutilize its potential.
Loneliness and isolation are at an all-time high in the United States. However, they stem from a multitude of causes. An empathic connection with a peer who has lived the same experience has immense potential for healing and recovery in both the PSW and the service user. The 988 Lifeline should include an integrated peer support service for both mental health and a broad range of lived experiences. While there exist some peer support services for conditions not related to mental health, these are not standardized or easily accessible to the entire American population. Many peer-operated warmlines (i.e., support lines) exist. However, they are spread across dozens of phone numbers and websites with varied and limited sources of funding. A single peer support service integrated into the 988 Lifeline would make peer support universally available. This service would address the emotional toll that accompanies a wide range of challenging life circumstances.
In short, a peer support service integrated into the 988 Lifeline would make the efforts of existing peer support organizations even more effective as it would:
- Standardize training, quality of care, and confidentiality,
- Expand the kinds of lived experience that are available for peer support, and
- Maximize the distribution of high-quality services across the U.S., with a focus on reaching high-risk populations.
Action Plan
The peer support service should begin by covering only a few kinds of lived experiences. After this implementation, the service should be expanded to cover a broader range of experiences.
Recommendation 1. Create a Peer Support Task Force (PSTF).
A PSTF should be established within SAMHSA. The secretary of the U.S. Department of Health and Human Services (HHS) should work with SAMHSA’s assistant secretary for mental health and substance use to establish this temporary task force.
The PSTF should lead the implementation of the peer support service, acting as an interagency task force that coordinates with partners across the public, private, and nonprofit sectors. This partnership will ensure that different lived experiences are accounted for and that existing resources are used effectively. The PSTF should collaborate with federal agencies, including the Department of Veterans Affairs (VA) and the Indian Health Service (IHS), as well as with advocacy organizations like NAMI and the American Cancer Society that champion the needs of people with specific lived experiences.
The PSTF should be charged with the following recommendations to start.
Recommendation 2. Integrate a peer support option into the 988 Lifeline.
Under the authority of the PSTF, integrating a peer support option into the 988 Lifeline could bypass additional Congressional action.
In the first pass, the integrated peer support option should cover only a few kinds of lived experiences (e.g., suicide and behavioral crises, veteran-connected disability).
- Before scaling the peer support service across the United States, the PSTF should implement a pilot program to test and refine protocols. The PSTF should select 988 Lifeline centers in states that already have a strong peer support program, and it should pilot peer support call and text services. The pilot program should select a few peer support needs to test first (e.g., suicide and behavioral crises, veteran-connected disability), as a narrow scope will be easier to assess for the first pass.
- The PSTF should then incorporate feedback from this pilot program as it scales up and integrates a peer support option into the nationwide 988 Lifeline.
- The PSTF should coordinate with the Federal Communications Commission (FCC) to add a caller menu option for peer support into the 988 Lifeline. (E.g., “Press 4 for peer support.”)
- The PSTF should coordinate with the FCC and U.S. Digital Service to implement a telephone triage, so the service user can submit a request for a specific kind of peer support and then be routed to a PSW on shift who has a lived experience that best matches the submitted request.
- The PSTF should facilitate the 988 Lifeline’s partnership with existing local and national peer support organizations and warmlines. Partnering with these existing organizations would bolster the 988 Lifeline’s capacity to provide peer support. Furthermore, states that already have a strong peer support certification in place could easily integrate their trained PSWs into the 988 Lifeline services using their existing infrastructure and expertise.
- The PSTF should coordinate with the FCC to incorporate a peer support text messaging option within the 988 Lifeline’s text and chat services. (For example, service users could text “PEER” to 9-8-8 and be routed to a PSW on shift.)
- The PSTF should implement a public campaign to the general public clarifying that the 988 Lifeline will remain as a suicide and crisis hotline, but it will also provide access to broader peer support services. The available peer support services should be clearly outlined on the 988 Lifeline website.
PSWs should work in call centers alongside 988 Lifeline phone and text specialists.
Recommendation 3. Develop an action plan to fund and sustain the integration of the peer support service.
The peer support service would need funding to become integrated into the 988 Lifeline. The government has recently approved funds for mental health initiatives, such as with the Restoring Hope for Mental Health and Well-Being Act of 2022. In 2023, HHS announced an additional $200 million in funding for the 988 Lifeline, but overall, HHS has granted almost $1.5 billion in total toward the 988 Lifeline. Similar avenues of funding should help jumpstart the integration of the peer support service.
For continued maintenance, fees for the peer support service should apply in a similar fashion to 9-1-1. That is, service users should not pay each time they access the 988 Lifeline or its integrated peer support service. Some U.S. states have already passed 9-8-8 implementation legislation that allows a monthly flat fee to be collected through telephone and wireless service providers, as permitted in the “National Suicide Hotline Designation Act of 2020”. The remaining states should be encouraged to pass similar legislation. The fee should remain in an account that is spent only for the maintenance of the 988 Lifeline and peer support services. If a fee is collected, then the FCC should provide an annual report on these fees and their usage.
Funding for the peer support service’s integration into the 988 Lifeline would entail the following:
- Maintenance of the peer support service
- Financial compensation for trainers who will train and mentor the PSWs
- Recruitment of trainers and PSWs
- The development of training resources for PSWs
Recommendation 4. Establish state-level standardized peer support training and certification.
PSWs should learn skills that are commonly taught to 988 Lifeline specialists, including active listening and recovery-oriented language. They should learn how to share their own stories, navigate challenging conversations, maintain boundaries, and self-care. Their training should be standardized at the state level, and it should be based on the “National Model Standards for Peer Support Certification” established by SAMHSA.
While most U.S. states have some version of peer support certification for mental health and/or substance use recovery, the PSTF should work with advocacy organizations to ensure that the state-level standardized training accounts for the peer support needs and demographics of each state. The peer support training should address the needs of people with a diverse range of lived experiences. Peer support needs can also be studied using caller outcome data that are recorded by 988 Lifeline specialists.
The PSTF should recruit trainers who will train and mentor the PSWs. To start, the PSTF should liaise within SAMHSA and with existing peer support organizations to coordinate this recruitment.
Recommendation 5. Establish a nation-wide system for the recruitment and training of PSWs.
Recruit individuals who would like to use their own lived experience to help other community members. Recruitment should occur through hospitals, clinics, as well as peer-run and community-based organizations to maximize recruitment pathways.
Current 988 Lifeline phone and text specialists could also be good candidates for the peer support service. Many are motivated by their own lived experiences and are already trained to handle calls across multiple helplines. 988 Lifeline specialists are instructed to focus on the service user seeking help and not share about themselves. However, specialists’ own lived experiences––if they are comfortable sharing them––represent untapped potential.
Once recruited, individuals who are training to become PSWs should attend live training sessions online with a peer cohort.
Recommendation 6. Expand the peer support service to cover a diverse range of lived experiences.
After successfully establishing the peer support service, it should be expanded upon to provide support for a broad and diverse range of lived experiences. The available peer support services should continue to be updated and outlined on the 988 Lifeline website.
Conclusion
A peer support service should be integrated into the 988 Lifeline. A service that caters to all kinds of lived experience paves way for a more empowered and self-reliant community. It fosters a societal mindset of helping each other based on shared lived experience in an empathic and healthy way. Peer support empowers both the PSW and the service user. While the service user finds empathy and understanding, the PSW finds a renewed sense of purpose and confidence. In this way, peer support would alleviate loneliness and isolation that result from a variety of causes while instilling longer-term resilience into the community.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
A service user refers to any individual who uses the peer support service for assistance.
Peer support work can be empowering and healing for those in recovery. However, as with any emotionally challenging work, PSWs benefit from ongoing support and supervision. Some interactions can be draining, especially if they hit too close to home. It is common for 988 Lifeline call centers to have a trained staff member, such as a psychologist or therapist, on call for such situations. A PSW could use this resource to debrief or process as needed. Additionally, during recruitment, PSW candidates should be screened to ensure they are comfortable speaking about their own lived experience and helping others who are going through the same experience.
Most PSWs would be compensated in the same way as their 988 Lifeline call and text specialist counterparts. They would be compensated with rigorous ongoing training, support, and resources for recovery and self-care in return for their time. They would also benefit from a social support system of fellow PSWs. Some individuals who may be ideal candidates for peer support work may struggle to find employment and health insurance. They will be less likely to volunteer if they do not have a living wage. To maximize the range of lived experiences available, certain individuals should be eligible for financial compensation.
A service user’s phone call or text should be routed to a 988 Lifeline peer support operator or telephone triage, where the service user is asked to say aloud (or type) what kind of support they are searching for (e.g., “I want to speak with someone who has been in prison.”) Then, the service user’s call (or text) would be routed to a PSW (under an alias) anywhere in the United States who 1) is currently on shift and 2) has a lived experience that is closest to the service user’s request. Each PSW would have associated keywords, such as “former incarceration,” “PTSD,” or “living alone,” which means that they are trained to connect with any service user about those lived experiences. The PSW would follow the service user’s lead in the conversation, and the PSW could share parts of their own lived experience when appropriate.
Text messaging can be more accessible than a phone line for youths and people with disabilities. The 988 Lifeline includes a text messaging option for this same reason.
It is true that both the regular 988 Lifeline and peer support service would provide resources and emotional support. By incorporating peer support into the 988 Lifeline, existing local and national peer support organizations would be eligible to partner with the 988 Lifeline and bolster its capacity to provide peer support. This endeavor will help with some staffing concerns. Furthermore, the peer support service would cover a diverse range of lived experiences extending beyond mental health. Therefore, more individuals may be motivated to join the 988 Lifeline staff to share their unique lived experience and help others who feel the same way.
Integrating a versatile peer support service into the 988 Lifeline transforms the latter into a service that every single American can use. (Every American has some unique lived experience.) The service’s versatility may help incentivize the remaining states to pass legislation to collect a 988 Lifeline fee through telephone and wireless service providers.
Finally, peer support is a cost-effective, preventative intervention. It should help remove the burden on other federal services, thereby reducing overall spending in time.
After implementing the action plan outlined in this memo, a subsequent memo should outline how to integrate peer support work into the community in person and on a large scale. Incorporating peer support into the 988 Lifeline would show its effectiveness to the public, policymakers, and healthcare professionals. This credibility would bolster endeavors to integrate peer support work into community settings throughout the country (e.g., behavioral health centers, hospitals and emergency rooms, and community clinics). These endeavors could also lead to professionalizing peer support, so PSWs can be reimbursed through Medicaid programs and health insurance.