A National Blueprint for Whole Health Transformation
Despite spending over 17% of GDP on health care, Americans live shorter and less healthy lives than their peers in other high-income countries. Rising chronic disease and mental health challenges as well as clinician burnout expose the limits of a system built to treat illness rather than create health. Addressing chronic disease while controlling healthcare costs is a bipartisan goal, the question now is how to achieve this shared goal? A policy window is opening now as Congress debates health care again – and in our view, it’s time for a “whole health” upgrade.
Whole Health is a proven, evidence-based framework that integrates medical care, behavioral health, public health, and community support so that people can live healthier, longer, and more meaningful lives. Pioneered by the Veterans Health Administration, Whole Health offers a redesign to U.S. health and social systems: it organizes how health is created and supported across sectors, shifting power and responsibility from institutions to people and communities. It begins with what matters most to people–their purpose, aspirations, and connections–and aligns prevention, clinical care, and social supports accordingly. Treating Whole Health as a shared public priority would help ensure that every community has the conditions to thrive.
Challenge and Opportunity
The U.S. health system spends over $4 trillion annually, more per capita than any other nation, yet underperforms on life expectancy, infant mortality, and chronic disease management. The prevailing fee-for-service model fragments care across medical, behavioral, and social domains, rewarding treatment over prevention. This fragmentation drives costs upward, fuels clinician burnout, and leaves many communities without coordinated support.
At this inflection point in our declining health outcomes and growing public awareness of the failures of our health system, federal prevention and public health programs are under review, governors are seeking cost-effective chronic disease solutions, and the National Academies is advocating for new healthcare models. Additionally, public demand for evidence-based well-being is growing, with 65% of Americans prioritizing mental and social health. There is clear demand for transformation in our health care system to deliver results in a much more efficient and cost effective way.
Veterans Health Administration’s Whole Health System Debuted in 2011
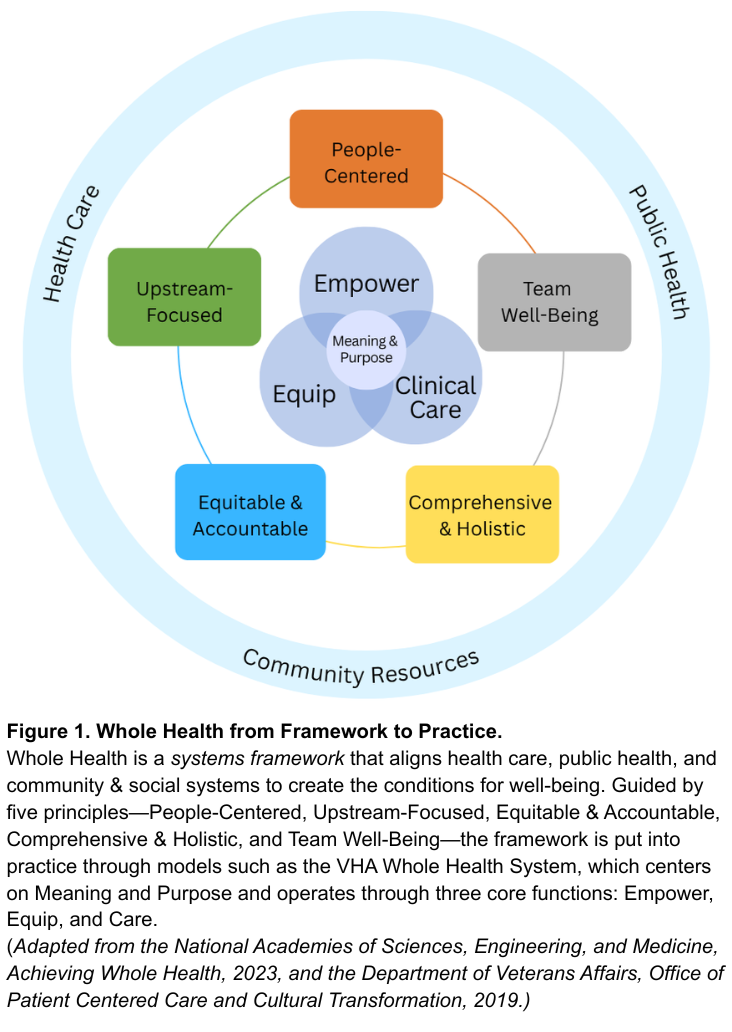
Whole Health offers a system-wide redesign for the challenge at hand. As defined by the National Academies of Sciences, Engineering, and Medicine, Whole Health is a framework for organizing how health is created and supported across sectors. It integrates medical care, behavioral health, public health, and community resources. As shown in Figure 1, the framework connects five system principles—People-Centered, Upstream-Focused, Equitable & Accountable, Comprehensive & Holistic, and Team Well-Being–that guide implementation across health and social support systems. The nation’s largest health system, the Veterans Health Administration’s (VHA), has demonstrated this framework in clinical practice through their Whole Health System since 2011. The VHA’s Whole Health System operates through three core functions: Empower (helping individuals define purpose), Equip (providing community resources like peer support), and Clinical Care (delivering coordinated, team-based care). Together, these elements align with what matters most to people, shifting the locus of control from expert-driven systems to shared agency through partnerships. The Whole Health System at the VHA has reduced opioid use and improved chronic disease outcomes.
Successful State Examples
Beyond the VHA, states have also demonstrated the possibility and benefits of Whole Health models. North Carolina’s Healthy Opportunities Pilots extended Medicaid coverage to housing, food, and transportation, showing fewer emergency visits and savings of about $85 per member per month. Vermont’s Blueprint for Health links primary care practices with community health teams and social services, reducing expenditures by about $480 per person annually and boosting preventive screenings. Finally, the Program of All-Inclusive Care for the Elderly (PACE), currently being implemented in 33 states, utilizes both Medicare and Medicaid funding to coordinate medical and social care for older adults with complex medical needs. While improvements can be made to national program-wide evaluation, states like Kansas have done evaluations that have found that the PACE program is less expensive than nursing homes per beneficiary and that nursing home admissions decline by 5% to 15% for beneficiaries.
Success across each of these examples relies on three pillars: (1) integrating medical, behavioral, social, and public health resources; (2) sustainable financing that prioritizes prevention and coordination; and (3) rigorous evaluation of outcomes that matter to people and communities. While these programs are early signs of success of Whole Health models, without coordinated leadership, efforts will fragment into isolated pilots and it will be challenging to learn and evolve.
A policy window for rethinking the health care system is opening. At this national inflection point, the U.S. can work to build a unified Whole Health strategy that enables a more effective, affordable and resilient health system.
Plan of Action
To act on this opportunity, federal and state leaders can take the following coordinated actions to embed Whole Health as a unifying framework across health, social, and wellbeing systems.
Recommendation 1. Declare Whole Health a Federal and State Priority.
Whole Health should become a unifying value across federal and state government action on health and wellbeing, embedding prevention, connection, and integration into how health and social systems are organized, financed, and delivered. Actions include:
- Federal Executive Action. The Executive Office of the President should create a Whole Health Strategic Council that brings together Veterans Affairs (VA), Health and Human Services (e.g. Centers for Disease Control and Prevention, Centers for Medicare and Medicaid (CMS), and Health Resources and Services Administration (HRSA)), Housing and Urban Development (HUD), and the U.S. Department of Agriculture (USDA) to align strategies, budgets, and programs with Whole Health principles through cross-agency guidance and joint planning. This council should also work with Governors to establish evidence-based benchmarks for Whole Health operations and evaluation (e.g., person-centered planning, peer support, team integration) and shared outcome metrics for well-being and population health.
- U.S. Congressional Action. Authorize whole health benefits, like housing assistance, nutrition counseling, transportation to appointments, peer support programs, and well-being centers as reimbursable services under Medicare, Medicaid and the Affordable Care Act health subsidies.
- State Action. Adopt Whole Health models through Medicaid managed-care contracts and through CDC and HRSA grant implementation. States should also develop support for Whole Health services in trusted local settings such as libraries, faith-based organizations, senior centers, to reach people where they live and gather.
Recommendation 2. Realign Financing and Payment to Reward Prevention and Team-Based Care.
Federal payment modalities need to shift from a fee-for-service model toward hybrid value-based models. Models such as per-member-per-month payments with quality incentives, can sustain comprehensive, team-based care while delivering outcomes that matter, like reductions in chronic disease and overall perceived wellbeing. Actions include:
- Federal Executive Action. Expand Advanced Primary Care Management (APCM) payments to include Whole Health teams, including clinicians, peer coaches, and community health workers. Ensure that this funding supports coordination, person-centered planning, and upstream prevention, such as food as medicine programs. Further, CMS can expand reimbursements to community health workers and peer support roles and standardize their scope-of-practice rules across states.
- U.S. Congressional Action. Invest in Medicare and Medicaid innovation programs, such as the CMS Innovation Center (CMMI), that reward prevention and chronic disease reduction. Additionally, expand tools for payment flexibility, through Medicaid waivers and state innovation funds, to help states adapt Whole Health models to local needs.
- State Action. Require Medicaid managed-care contracts to reimburse Whole Health services, particularly in underserved and rural areas, and encourage payers to align benefit designs and performance measures around well-being. States should also leverage their state insurance departments to guide and incentivize private health insurers to adopt Whole Health payment models.
Recommendation 3. Strengthen and Expand the Whole Health Workforce.
Whole Health practice needs a broad team to be successful: clinicians, community health workers, peer coaches, community organizations, nutritionists, and educators. To build this workforce, governments need to modernize training, assess the workforce and workplace quality, and connect the fast-growing well-being sector with health and community systems. Actions include:
- Federal Executive Action. Through VA and HRSA establish Whole Health Workforce Centers of Excellence to develop national curricula, set standards, and disseminate evidence on effective Whole Health team-building. Further, CMS should track workforce outcomes such as retention, burnout, and team integration, and evaluate the benefits for health professionals working in Whole Health systems versus traditional health systems.
- U.S. Congressional Action. Expand CMS Graduate Medical Education Funds and HRSA workforce programs to support Whole Health training, certifications, and placements across clinical and community settings.
- State Action. As a part of initiatives to grow the health workforce, state governments should expand the definition of a “health professional” to include Whole Health practitioners. Further, states can leverage their role as a licensure for professionals by creating a “whole health” licensing process that recognizes professionals that meet evidence-based standards for Whole Health.
Recommendation 4. Build a National Learning and Research Infrastructure.
Whole Health programs across the country are proving effective, but lessons remain siloed. A coordinated national system should link evidence, evaluation, and implementation so that successful models can scale quickly and sustainably.
- Federal Executive Action. Direct the Agency for Healthcare Research and Quality, National Institutes of Health, and partner agencies (VA, HUD, USDA) to run pragmatic trials and cost-effectiveness studies of Whole Health interventions that measure well-being across clinical, biomedical, behavioral, and social domains. The federal government should also embed Whole Health frameworks into government-wide research agendas to sustain a culture of evidence-based improvement.
- U.S. Congressional Action. Charter a quasi-governmental entity, modeled on Patient-Centered Outcomes Research Institute (PCORI), to coordinate Whole Health demonstration sites and research. This new entity should partner with CMMI, HRSA and VA to test Whole Health payment and delivery models under real-world conditions. This entity should also establish an interagency team as well as state network to address payment, regulatory, and privacy barriers identified by sites and pilots.
- State Action. Partner with federal agencies through innovation waivers (e.g. 1115 waivers and 1332 waivers) and learning collaboratives to test Whole Health models and share data across state systems and with the federal government.
Conclusion
The United States spends more on health care than any other nation yet delivers poorer outcomes. Whole Health offers a proven path to reverse this trend, reframing care around prevention, purpose, and integration across health and social systems. Embedding Whole Health as the operating system for America’s health requires three shifts: (1) redefining the purpose from treating disease to optimizing health and well-being; (2) restructuring care to empower, equip, and treat through team-based and community-linked approaches; and (3) rebalancing control from expert-driven systems to partnerships guided by what matters most to people and communities. Federal and state leaders have the opportunity to turn scattered Whole Health pilots to a coordinated national strategy. The cost of inaction is continued fragmentation; the reward of action is a healthier and more resilient nation.
This memo produced as part of Strengthening Pathways to Disease Prevention and Improved Health Outcomes.
Both approaches emphasize caring for people as integrated beings rather than as a collection of diseases, but they differ in scope and application. Whole Person Health, as used by NIH, focuses on the biological, psychological, and behavioral systems within an individual—it is primarily a research framework for understanding health across body systems. Whole Health is a systems framework that extends beyond the individual to include families, communities, and environments. It integrates medical care, behavioral health, public health, and social support around what matters most to each person. In short, Whole Person Health is about how the body and mind work together; Whole Health is about how health, social, and community systems work together to create the conditions for well-being. Policymakers can use Whole Health to guide financing, workforce, and infrastructure reforms that translate Whole Person Health science into everyday practice.
Integrative Health combines evidence-based conventional and complementary approaches such as mindfulness, acupuncture, yoga, and nutrition to support healing of the whole person. Whole Health extends further. It includes prevention, self-care, and personal agency, and moves beyond the clinic to connect medical care with social, behavioral, and community dimensions of health. Whole Health uses integrative approaches when evidence supports them, but it is ultimately a systems model that aligns health, social, and community supports around what matters most to people. For policymakers, it provides a structure for integrating clinical and community services within financing and workforce strategies.
They share a common foundation but differ in scope and audience. The VA Whole Health System, developed by the Department of Veterans Affairs, is an operational model, a way of delivering care that helps veterans identify what matters most, supports self-care and skill building, and provides team-based clinical treatment. The National Academies’ Whole Health framework builds on the VA’s experience and expands it to the national level. It is a policy and systems framework that applies Whole Health principles across all populations and connects health care with public health, behavioral health, and community systems. In short, the VA model shows how Whole Health works in practice, while the National Academies framework shows how it can guide national policy and system alignment.
In Honor of Patient Safety Day, Four Recommendations to Improve Healthcare Outcomes
Through partnership with the Doris Duke Foundation, FAS is working to ensure that rigorous, evidence-based ideas on the cutting edge of disease prevention and health outcomes are reaching decision makers in an effective and timely manner. To that end, we have been collaborating with the Strengthening Pathways effort, a series of national conversations held in spring 2025 to surface research questions, incentives, and overlooked opportunities for innovation with potential to prevent disease and improve outcomes of care in the United States. FAS is leveraging its skills in policy entrepreneurship, working with session organizers, to ensure that ideas surfaced in these symposia reach decision-makers to drive impact in active policy windows.
On this World Patient Safety Day 2025, we share a set of recommendations that align with the National Quality Strategy of Centers for Medicare and Medicaid Services (CMS) goal for zero preventable harm in healthcare. Working with Patients for Patient Safety US, which co-led one of Strengthening Pathways conversations this spring with the Johns Hopkins University Armstrong Institute for Patient Safety and Quality, the issue brief below outlines a bold, modernized approach that uses Artificial Intelligence technology to empower patients and drive change. FAS continues to explore the rapidly evolving AI and healthcare nexus.
Patient safety is an often-overlooked challenge in our healthcare systems. Whether safety events are caused by medical error, missed or delayed diagnoses, deviations from standards of care, or neglect, hundreds of billions of dollars and hundreds of thousands of lives are lost each year due to patient safety lapses in our healthcare settings. But most patient safety challenges are not really captured and there are not enough tools to empower clinicians to improve. Here we present four critical proposals for improving patient safety that are worthy of attention and action.
Challenge and Opportunity
Reducing patient death and harm from medical error surfaced as a U.S. public health priority at the turn of the century with the landmark National Academy of Sciences (NAS) report, To Err is Human: Building a Safer Health System (2000). Research shows that medical error is the 3rd largest cause of preventable death in the U.S. Analysis of Medicare claims data and electronic health records by the Department of Health and Human Services (DHHS) Office of the Inspector General (OIG) in a series of reports from 2008 to 2025 consistently finds that 25-30% of Medicare recipients experience harm events across multiple healthcare settings, from hospitals to skilled nursing facilities to long term care hospitals to rehab centers. Research on the broader population finds similar rates for adult patients in hospitals. The most recent study on preventable harm in ambulatory care found that 7% of patients experienced at least one adverse event, with wide variation of 1.8% to 23.6% from clinical setting to clinical setting. Improving diagnostic safety has emerged as the largest opportunity for patient harm prevention. New research estimates 795,000 patients in the U.S. annually experience death or harm due to missed, delayed or ineffectively communicated diagnoses. The annual cost to the health care system of preventable harm and its health care cascades is conservatively estimated to exceed $200 billion. This cost is ultimately borne by families and taxpayers.
In its National Quality Strategy, the Centers for Medicare and Medicaid Services (CMS) articulated an aspirational goal of zero preventable harm in healthcare. The National Action Alliance for Patient and Workforce Safety, now managed by the Agency for Healthcare Research and Quality (AHRQ), has a goal of 50% reduction in preventable harm by 2026. These goals cannot be achieved without a bold, modernized approach that uses AI technology to empower patients and drive change. Under-reporting negative outcomes and patient harms keeps clinicians and staff from identifying and implementing solutions to improve care. In its latest analysis (July 2025), the OIG finds that fewer than 5% of medical errors are ever reported to the systems designed to gather insights from them. Hospitals failed to capture half of harm events identified via medical record review, and even among captured events, few led to investigation or safety improvements. Only 16% of events required to be reported externally to CMS or State entities were actually reported, meaning critical oversight systems are missing safety signals entirely.
Multiple research papers over the last 20 years find that patients will report things that providers do not. But there has been no simple, trusted way for patient observations to reach the right people at the right time in a way that supports learning and Improvement. Patients could be especially effective in reporting missed or delayed diagnoses, which often manifest across the continuum of care, not in one healthcare setting or a single patient visit. The advent of AI systems provides an unprecedented opportunity to address patient safety and improve patient outcomes if we can improve the data available on the frequency and nature of medical errors. Here we present four ideas for improving patient safety.
Recommendation 1. Create AI-Empowered Safety Event Reporting and Learning System With and For Patients
The Department of Health and Human Services (HHS) can, through CMS, AHRQ or another HHS agency, develop an AI-empowered National Patient Safety Learning and Reporting System that enables anyone, including patients and families, to directly report harm events or flag safety concerns for improvement, including in real or near real time. Doing so would make sure everyone in the system has the full picture — so healthcare providers can act quickly, learn faster, and protect more patients.
This system will:
- Develop a reporting portal to collect, triage and analyze patient reported data directly from beneficiaries to improve patient and diagnostic safety.
- Redesign and modernize Consumer Assessment of Healthcare Providers and Systems
(CAHPS) surveys to include questions that capture beneficiaries’ experiences and outcomes related to patient and diagnostic safety events.
- Redefine the Beneficiary and Family Centered Care Quality Improvement Organizations (BFCC QIO) scope of work to integrate the QIOs into the National Patient Safety Learning and Reporting System.
The learning system will:
- Use advanced triage (including AI) to distinguish high-signal events and route credible
reports directly to the care team and oversight bodies that can act on them.
- Solicit timely feedback and insights in support of hospitals, clinics, and nursing homes to prevent recurrence, as well as feedback over time on patient outcomes that manifest later, e.g. as a result of missed or delayed diagnoses.
- Protect patients and providers by focusing on efficacy of solutions, not blame assignment.
- Feed anonymized, interoperable data into a national learning network that will spot systemic risks sooner and make aggregated data available for transparency and system learning.
Recommendation 2. Create a Real-time ‘Patient Safety Dashboard’ using AI
HHS should build an AI-driven platform that integrates patient-reported safety data — including data from the new National Patient Reporting and Learning System, recommended above — with clinical data from electronic health records to create a real-time ‘patient safety dashboard’ for hospitals and clinics. This dashboard will empower providers to improve care in real time, and will:
- Assist health care providers make accurate and timely diagnoses and avoid errors.
- Make patient reporting easy, effective, and actionable.
- Use AI to triage harm signals and detect systemic risk in real time.
- Build shared national infrastructure for healthcare reporting for all stakeholders.
- Align incentives to reward harm reduction and safety.
By harnessing the power of AI providers will be able to respond faster, identify patients at risk more effectively, and prevent harm thereby improving outcomes. This “central nervous system” for patient safety will be deployed nationally to help detect safety signals in real time, connect information across settings, and alert teams before harm occurs.
Recommendation 3. Mine Billing Data for Deviations from Standards of Care
Standards of care are guidelines that define the process, procedures and treatments that patients should receive in various medical and professional contexts. Standards ensure that individuals receive appropriate and effective care based on established practices. Most standards of care are developed and promulgated by medical societies. But not all clinicians and clinical settings adhere to standards of care, and deviations from standards of care are normal depending upon the case before them. Nonetheless, standards of care exist for a reason and deviations from standards of care should be noted when medical errors result in negative outcomes for patients so that clinicians can learn from these outcomes and improve.
Some patient safety challenges are evident right in the billing data submitted to CMS and insurers. For example, deviations from standards of care can be captured in billing data by comparing clinical diagnosis codes with billing codes and then compared to widely accepted standards of care. By using CMS billing data, the government could identify opportunities for driving the development, augmentation, and wider adoption of standards of care by showing variability and compliance with standards of care for patients, reducing medical error and improving outcomes.
Giving standard setters real data to adapt and develop new standards of care is a powerful tool for improving patient outcomes.
Recommendation 4. Create a Patient Safety AI Testbed
HHS can also establish a Patient Safety AI Testbed to evaluate how AI tools used in diagnosis, monitoring, and care coordination perform in real-world settings. This testbed will ensure that AI improves safety, not just efficiency — and can be co-led by patients, clinicians, and independent safety experts. This is an expansion of the testbeds in the HHS AI Strategic Plan.
The Patient Safety Testbed could include:
- Funding for independent AI test environments to monitor real-world safety and performance over time.
- Public reliability benchmarks and “AI safety labeling”.
- Required participation by AI vendors and provider systems.
Conclusion
There are several key steps that the government can take to address the major loss of health, dollars, and lives due to medical errors, while simultaneously bolstering treatment guidelines, driving the development of new transparent data, and holding the medical establishment accountable for improving care. Here we present four proposals. None of them are particularly expensive when juxtaposed against the tremendous savings they will drive throughout our healthcare system. We can only hope that the Administration’s commitment to patient safety is such that they will adopt them and drive a new era where caregivers, healthcare systems and insurance payers work together to improve patient safety and care standards.
This memo produced as part of Strengthening Pathways to Disease Prevention and Improved Health Outcomes.
Terminal Patients Need Better Access to Drugs and Clinical Trial Information
Editor’s note: This policy memo was written by Jake Seliger and his wife Bess Stillman. Jake passed away before meeting his child, Athena, born on October 31, 2024. Except where indicated, the first-person voice is that of Jake. This memo advocates for user-centric technology modernization and data interoperability. More crucially, he urges expanded patient rights to opt-in to experimental drug trials and FDA rule revisions to enable terminal patients to take more risks on behalf of themselves, for the benefit of others.
The FDA is supposed to ensure that treatments are safe and effective, but terminal cancer patients like me are already effectively dead. If I don’t receive advanced treatment quickly, I will die. My hope, in the time I have remaining, is to promote policies that will speed up access to treatments and clinical trials for cancer patients throughout the U.S.
There are about two million cancer diagnoses and 600,000 deaths annually in the United States. Cancer treatments are improved over time via the clinical trial system: thousands of clinical trials are conducted each year (many funded by the government via the National Institutes of Health, or NIH, and many funded by pharmaceutical companies hoping to get FDA approval for their products).
But the clinical trial system is needlessly slow, and as discussed below, is nearly impossible for any layperson to access without skilled consulting. As a result, clinical trials are far less useful than they could be.
The FDA is currently “protecting” me from being harmed or killed by novel, promising, advanced cancer treatments that could save or extend my life, so that I can die from cancer instead. Like most patients, I would prefer a much faster system in which the FDA conditionally approves promising, early-phase advanced cancer treatments, even if those treatments haven’t yet been proven fully effective. Drugmakers will be better incentivized to invest in novel cancer treatments if they can begin receiving payment for those treatments sooner. The true risks to terminal patients like me are low—I’m already dying—and the benefits to both existing terminal patients and future patients of all kinds are substantial.
I would also prefer a clinical trial system that was easy for patients to navigate, rather than next-to-impossible. Easier access for patients could radically lower the cost and time for clinical trials by making recruitment far cheaper and more widespread, rather than including only about 6% of patients. In turn, speeding up the clinical-trial process means that future cancer patients will be helped by more quickly approving novel treatments. About half of pharmaceutical R&D spending goes not to basic research, but to the clinical trial process. If we can cut the costs of clinical trials by streamlining the process to improve access to terminal patients, more treatments will make it to patients, and will be faster in doing so.
Cancer treatment is a non-partisan issue. To my knowledge, both left and right agree that prematurely dying from cancer is bad. Excess safety-ism and excessive caution from the FDA costs lives, including in the near future, my own. Three concrete actions would improve clinical research, particular for terminal cancer patients like me, but for many other patients as well:
Clinical trials should be easier and cheaper. The chief obstacles to this are recruitment and retention.
Congress and NIH should modernize the website ClinicalTrials.gov and vastly expand what drug companies and research sites are required to report there, and the time in which they must report it. Requiring timely updates that include comprehensive eligibility criteria, availability for new participants, and accurate site contact information,would mean that patients and doctors will have much more complete information about what trials are available and for whom.
The process of determining patient eligibility and enrolling in a trial should be easier. Due to narrow eligibility criteria, studies struggle to enroll an adequate number of local patients, which severely delays trial progression. A patient who wishes to participate in a trial must “establish care” with the hospital system hosting the trial, before they are even initially screened for eligibility or told if a slot is available. Due to telemedicine practice restrictions across state lines, this means that patients who aren’t already cared for at that site— patients who are ill and for whom unnecessary travel is a huge burden— must use their limited energy to travel to a site just to find out if they can proceed to requesting a trial slot and starting further eligibility testing. Then, if approved for the study, they must be able to uproot their lives to move to, or spend extensive periods of time at, the study location
Improved access to remote care for clinical trials would solve both these problems. First, by allowing the practice of telemedicine across state lines for visits directly related to screening and enrollment into clinical trials. Second, by incentivizing decentralization—meaning a participant in the study can receive the experimental drug and most monitoring, labs and imaging at a local hospital or infusion clinic—by accepting data from sites that can follow a standardized study protocol.
We should require the FDA to allow companies with prospective treatments for fatal diseases to bring those treatments to market after initial safety studies, with minimal delays and with a lessened burden for demonstrating benefit.
Background
[At the time of writing this] I’m a 40-year-old man whose wife is five months pregnant, and the treatment that may keep me alive long enough to meet my child is being kept from me because of current FDA policies. That drug may be in a clinical trial I cannot access. Or, it may be blocked from coming to market by requirements for additional testing to further prove efficacy that has already been demonstrated.
Instead of giving me a chance to take calculated risks on a new therapy that might allow me to live and to be with my family, current FDA regulations are choosing for me: deciding that my certain death from cancer is somehow less harmful to me than taking a calculated, informed risk that might save or prolong my life. Who is asking the patients being protected what they would rather be protected from? The FDA errs too much on the side of extreme caution around drug safety and efficacy, and that choice leads to preventable deaths.
One group of scholars attempted to model how many lives are lost versus gained from a more or less conservative FDA. They find that “from the patient’s perspective, the approval criteria in [FDA program accelerators] may still seem far too conservative.” Their analysis is consistent with the FDA being too stringent and slow in approving use of drugs for fatal diseases like cancer: “Our findings suggest that conventional standards of statistical significance for approving drugs may be overly conservative for the most deadly diseases.”
Drugmakers also find it difficult to navigate what exactly the FDA wants: “their deliberations are largely opaque—even to industry insiders—and the exact role and weight of patient preferences are unclear.” This exacerbates the difficulty drugmakers face in seeking to get treatments to patients faster. Inaction in the form of delaying patient access to drugs is killing people. Inaction is choosing death for cancer patients.
I’m an example of this; I was diagnosed with squamous cell carcinoma (SCC) of the tongue in Oct. 2022. I had no risk factors, like smoking or alcoholism, that put me at risk for SCC. The original tumor was removed in October 2022, and I then had radiation that was supposed to cure me. It didn’t, and the cancer reappeared in April 2023. At that point, I would’ve been a great candidate for a drug like MCLA-158, which has been stuck in clinical trials, despite “breakthrough therapy designation” by the FDA and impressive data, for more than five years. This, despite the fact that MCLA-158 is much easier to tolerate than chemotherapy and arrests cancer in about 70% of patients. Current standard of care chemotherapy and immunotherapy has a positive response rate of only 20-30%.
Had MCLA-158 been approved, I might have received it in April 2023, and still have my tongue. Instead, in May 2023, my entire tongue was surgically removed in an attempt to save my life, forever altering my ability to speak and eat and live a normal life. That surgery removed the cancer, but two months later it recurred again in July 2023. While clinical-trial drugs are keeping me alive right now, I’m dying in part because promising treatments like MCLA-158 are stuck in clinical trials, and I couldn’t get them in a timely fashion, despite early data showing their efficacy. Merus, the maker of MCLA-158, is planning a phase 3 trial for MCLA-158, despite its initial successes. This is crazy: head and neck cancer patients need MCLA-158 now.

I’m only writing this brief because I was one of the few patients who was indeed, at long last, able to access MCLA-158 in a clinical trial, which incredibly halted my rapidly expanding, aggressive tumors. Without it, I’d have been dead nine months ago. Without it, many other patients already are. Imagine that you, or your spouse, or parent, or child, finds him or herself in a situation like mine. Do you want the FDA to keep testing a drug that’s already been shown to be effective and allow patients who could benefit to suffer and die? Or do you want your loved one to get the drug, and avoid surgical mutilation and death? I know what I’d choose.
Multiply this situation across hundreds of thousands of people per year and you’ll understand the frustration of the dying cancer patients like me.
As noted above, about 600,000 people die annually from cancer—and yet cancer drugs routinely take a decade or more to move from lab to approval. The process is slow: “By the time a drug gets into phase III, the work required to bring it to that point may have consumed half a decade, or longer, and tens if not hundreds of millions of dollars.” If you have a fatal disease today, a treatment that is five to ten years away won’t help. Too few people participate in clinical trials partly because participation is so difficult; one study finds that “across the entire U.S. system, the estimated participation rate to cancer treatment trials was 6.3%.” Given how many people die, the prospect of life is attractive.
There is another option in between waiting decades for a promising drug to come to market and opening the market to untested drugs: Allowing terminal patients to consent to the risk of novel, earlier-phase treatments, instead of defaulting to near-certain death, would potentially benefit those patients as well as generate larger volumes of important data regarding drug safety and efficacy, thus improving the speed of drug approval for future patients. Requiring basic safety data is reasonable, but requiring complete phase 2 and 3 data for terminal cancer patients is unreasonably slow, and results in deaths that could be prevented through faster approval.
Again, imagine you, your spouse, parent, or child, is in a situation like mine: they’ve exhausted current standard-of-care cancer treatments and are consequently facing certain death. New treatments that could extend or even save their life may exist, or be on the verge of existing, but are held up by the FDA’s requirements that treatments be redundantly proven to be safe and highly effective. Do you want your family member to risk unproven but potentially effective treatments, or do you want your family member to die?
I’d make the same choice. The FDA stands in the way.
Equally important, we need to shorten the clinical trial process: As Alex Telford notes, “Clinical trials are expensive because they are complex, bureaucratic, and reliant on highly skilled labour. Trials now cost as much as $100,000 per patient to run, and sometimes up to $300,000 or even $500,000 per patient.” And as noted above, about half of pharmaceutical R&D spending goes not to basic research, but to the clinical trial process.
Cut the costs of clinical trials, and more treatments will make it to patients, faster. And while it’s not reasonable to treat humans like animal models, a lot of us who have fatal diagnoses have very little to lose and consequently want to try drugs that may help us, and people in the future with similar diseases. Most importantly, we understand the risks of potentially dying from a drug that might help us and generate important data, versus waiting to certainly die from cancer in a way that will not benefit anybody. We are capable of, and willing to give, informed consent. We can do better and move faster than we are now. In the grand scheme of things, “When it comes to clinical trials, we should aim to make them both cheaper and faster. There is as of yet no substitute for human subjects, especially for the complex diseases that are the biggest killers of our time. The best model of a human is (still) a human.” Inaction will lead to the continued deaths of hundreds of thousands of people annually.
Trials need patients, but the process of searching for a trial in which to enroll is archaic. We found ClinicalTrials.gov nearly impossible to navigate. Despite the stakes, from the patient’s perspective, the clinical trial process is impressively broken, obtuse and confusing, and one that we gather no one likes: patients don’t, their families don’t, hospitals and oncologists who run the clinical trials don’t, drug companies must not, and the people who die while waiting to get into a trial probably don’t.
I [Bess] knew that a clinical trial was Jake’s only chance. But how would we find one? I’d initially hoped that a head and neck oncologist would recommend a specific trial, preferably one that they could refer us to. But most doctors didn’t know of trial options outside their institution, or, frequently, within it, unless they were directly involved. ,Most recommended large research institutions that had a good reputation for hard cases, assuming they’d have more studies and one might be a match.
How were they, or we, supposed to find out what trials actually existed?
The only large-scale search option is ClinicalTrials.gov. But many oncologists I spoke with don’t engage with ClinicalTrials.gov, because the information is out-of-date, difficult to navigate, and inaccurate. It can’t be relied on. For example, I shared a summary of Jake’s relevant medical information (with his permission) in a group of physicians who had offered to help us with the clinical trial search. Ten physicians shared their top-five search results. Ominously, none listed the same trials.
How is it that ten doctors can put in the same basic, relevant clinical data into an engine meant to list and search for all existing clinical trials, only for no two to surface the same study? The problem is simple: There’s a lack of keyword standardization.
Instead of a drop-down menu or click-boxes with diagnoses to choose from, the first search “filter” on ClinicalTrials.gov is a text box that says “Condition\Disease.” If I search: “Head and Neck Cancer” I get ___________ results. If I search “Tongue Cancer,” I get _________ results. Although Tongue Cancer is a subset of Head and Neck Cancer, I don’t see the studies listed as “Head and Neck Cancer”, unless I type in both, or the person who created the ClinicalTrials.gov post for the study chose to type out multiple variations on a diagnosis. Nothing says they have to. If I search for both, I will still miss studies filed as: “HNSCC” or “All Solid Tumors” or “Squamous Cell Carcinoma of the Tongue.”
The good news is that online retailers solved this problem for us years ago. It’s easier to find a dress to my exact specifications out of thousands on H&M,com than it is to find a clinical trial. I can open a search bar, click “dress,” select my material from another click box (which allows me to select from the same options the people listing the garments chose from), then click on the boxes for my desired color, dry clean or machine wash, fabric, finish, closure, and any other number of pre-selected categories before adding additional search keywords if I choose. I find a handful of options all relevant to my desires within a matter of minutes. H&M provides a list of standardized keywords describing what they are offering, and I can filter from there. This way, H&M and I are speaking the same language. And a dress isn’t life or death. For much more on my difficulties with ClinicalTrials.gov, see here.
Further slowing a patient’s ability to find a relevant clinical trial is a lack of comprehensive, searchable, eligibility criteria. Every study has eligibility criteria, and eligibility criteria—like keywords—aren’t standardized on ClinicalTrials.gov. Nor is it required that an exhaustive explanation of eligibility criteria be provided, which may lead to a patient wasting precious weeks attempting to establish care and enroll in a trial, only to discover there was unpublished eligibility criteria they don’t meet. Instead, the information page for each study outlines inclusion and exclusion criteria using whatever language whoever is typing feels like using. Many have overlapping inclusion and exclusion criteria, but there can be long lists of additional criteria for each arm of a study, and it’s up to the patient or their doctor to read through them line by line—if they’re even listed— to see if prior medications, current medications, certain genomic sequencing findings, numbers of lines of therapy, etc. makes the trial relevant.
In the end, we hired a consultant (Eileen), who leveraged her full-time work helping pharmaceutical companies determine which novel compounds might be worth pouring their R&D efforts into assisting patients find potential clinical trials. She helped us narrow it down to the top 5 candidate trials from the thousands that turned up in initial queries.
- NCT04815720: Pepinemab in Combination With Pembrolizumab in Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-B84)
- NCT03526835: A Study of Bispecific Antibody MCLA-158 in Patients With Advanced Solid Tumors.
- NCT05743270: Study of RP3 in Combination With Nivolumab and Other Therapy in Patients With Locoregionally Advanced or Recurrent SCCHN.”
- NCT03485209: Efficacy and Safety Study of Tisotumab Vedotin for Patients With Solid Tumors (innovaTV 207)
- NCT05094336: AMG 193, Methylthioadenosine (MTA) Cooperative Protein Arginine Methyltransferase 5 (PRMT5) Inhibitor, Alone and in Combination With Docetaxel in Advanced Methylthioadenosine Phosphorylase (MTAP)-Null Solid Tumors (MTAP).
Based on the names alone, you can see why it would be difficult if not impossible for someone without some expertise in cancer oncology to evaluate trials. Even with Eileen’s expertise, two of the trials were stricken from our list when we discovered unlisted eligibility criteria, which excluded Jake. When exceptionally motivated patients, the oncologists who care for them, and even consultants selling highly specialized assistance can’t reliably navigate a system that claims to be desperate to enroll patients into trials, there is a fundamental problem with the system. But this is a mismatch we can solve, to everyone’s benefit.
Plan of Action
We propose three major actions. Congress should:
Recommendation 1. Direct the National Library of Medicine (NLM) at the National Institutes of Health (NIH) to modernize ClinicalTrials.gov so that patients and doctors have complete and relevant information about available trials, as well as requiring more regular updates from companies as to all the details of available trials, and
Recommendation 2. Allow the practice of telemedicine across state lines for visits related to clinical trials.
Recommendation 3. Require the FDA to allow companies with prospective treatments for fatal diseases to bring those treatments to market after initial safety studies.
Modernizing ClinicalTrials.gov will empower patients, oncologists, and others to better understand what trials are available, where they are available, and their up-to-date eligibility criteria, using standardized search categories to make them more easily discoverable. Allowing telemedicine across state lines for clinical trial care will significantly improve enrollment and retention. Bringing treatments to market after initial safety studies will speed the clinical trial process, and get more patients treatments, sooner. In cancer, delays cause death.
To get more specific:
The FDA already has a number of accelerated approval options. Instead of the usual “right to try” legislation, we propose creating a second, provisional market for terminal patients that allows partial approval of a drug while it’s still undergoing trials, making it available to trial-ineligible (or those unable to easily access a trial) patients for whom standard of care doesn’t provide a meaningful chance at remission. This partial approval would ideally allow physicians to prescribe the drug to this subset of patients as they see fit: be that monotherapy or a variety of personalized combination therapies, tailored to a patient’s needs, side-effect profile and goals. This wouldn’t just expand access to patients who are otherwise out of luck, but can give important data about real-world reaction and response.
As an incentive, the FDA could require pharmaceutical companies to provide drug access to terminal patients as a condition of continuing forward in the trial process on the road to a New Drug Application. While this would be federally forced compassion, it would differ from “compassionate use.” To currently access a study drug via compassionate use, a physician has to petition both the drug company and the FDA on the patient’s behalf, which comes with onerous rules and requirements. Most drug companies with compassionate use programs won’t offer a drug until there’s already a large amount of compelling phase 2 data demonstrating efficacy, a patient must have “failed” standard of care and other available treatments, and the drug must (usually) be given as a monotherapy. Even if the drugmaker says yes, the FDA can still say no. Compassionate use is an option available to very small numbers, in limited instances, and with bureaucratic barriers to overcome. Not terribly compassionate, in my opinion.
The benefits of this “terminal patient” market can and should go both ways, much as lovers should benefit from each other instead of trying to create win-lose situations. Providing the drug to patients would come with reciprocal benefits to the pharmaceutical companies. Any physician prescribing the drug to patients should be required to report data regarding how the drug was used, in what combination, and to what effect. This would create a large pool of observational, real-world data gathered from the patients who aren’t ideal candidates for trials, but better represent the large subset of patients who’ve exhausted multiple lines of therapies yet aren’t ready for the end. Promising combinations and unexpected effects might be identified from these observational data sets, and possibly used to design future trials.
Dying patients would get drugs faster, and live longer, healthier lives, if we can get better access to both information about clinical trials, and treatments for diseases. The current system errs too much on proving effectiveness and too little on the importance of speed itself, and of the number of people who die while waiting for new treatments. Patients like me, who have fatal diagnoses and have already failed “standard of care” therapies, routinely die while waiting for new or improved treatments. Moreover, patients like me have little to lose: because cancer is going to kill us anyway, many of us would prefer to roll the dice on an unproven treatment, or a treatment that has early, incomplete data showing its potential to help, than wait to be killed by cancer. Right now, however, the FDA does not consider how many patients will die while waiting for new treatments. Instead, the FDA requires that drugmakers prove both the safety and efficacy of treatments prior to allowing any approval whatsoever.
As for improving ClinicalTrials.gov, we have several ideas:
First, the NIH should hire programmers with UX experience, and should be empowered to pay market rates to software developers with experience at designing websites for, say, Amazon or Shein. Clinicaltrials.gov was designed to be a study registry, but needs an overhaul to be more useful to actual patients and doctors.
Second, UX is far from the only problem. Data itself can be a problem. For example, the NIH could require a patient-friendly summary of each trial; it could standardize the names of drugs and conditions so that patients and doctors could see consistent and complete search results; and it could use a consistent and machine-readable format for inclusion and exclusion criteria.
We are aware of one patient group that, in trying to build an interface to ClinicalTrials.gov, found a remarkable degree of inconsistency and chaos: “We have analyzed the inclusion and exclusion criteria for all the cancer-related trials from clinicaltrials.gov that are recruiting for interventional studies (approximately 8,500 trials). Among these trials, there are over 1,000 ways to indicate the patient must not be pregnant during a trial, and another 1,000 ways to indicate that the patient must use adequate birth control.” ClinicalTrials.gov should use a Domain Specific Language that standardizes all of these terms and conditions, so that patients and doctors can find relevant trials with 100x less effort.
Third, the long-run goal should be a real-time, searchable database that matches patients to studies using EMR data and allows doctors to see what spots are available and where. Pharmaceutical and biotech companies would need to be required to contribute up-to-date information on all extant clinical trials on a regular basis (e.g., monthly).
Conclusion
Indeed, we need a national clinical trial database that EMRs can connect to, in the style of Epic’s Care Everywhere. A patient signs a release to share their information, like Care Everywhere allows hospital A to download a patient’s information from hospital B if the patient signs a release. Trials with open slots could mark themselves as “recruiting” and update in real time. A doctor could press a button and identify potential open studies. A patient available for a trial could flag their EMR profile as actively searching, allowing clinical trial sites to browse patients in their region who are looking for a study. Access to that patient’s EMR would allow them to scan for eligibility.
This would be easy to do. OKCupid can do it. The tech exists. Finding a clinical trial already feels a bit like online dating, except if you get the wrong match, you die.
This memo produced as part of the Federation of American Scientists and Good Science Project sprint. Find more ideas at Good Science Project x FAS
The FDA has created a variety of programs that have promising-sounding names: “Currently, four programs—the fast track, breakthrough therapy, accelerated approval, and priority review designations—provide faster reviews and/or use surrogate endpoints to judge efficacy. However, published descriptions […] do not indicate any differences in the statistical thresholds used in these programs versus the standard approval process, nor do they mention adapting these thresholds to the severity of the disease.” The problem is that these programs do not appear to do much to actually accelerate getting drugs to patients. MCLA-158 is an example of the problem: the drug has been shown to be safe and effective, and yet Merus thinks it needs a Phase 3 trial to get it past the FDA and to patients.
Improve healthcare data capture at the source to build a learning health system
Studies estimate that only one in 10 recommendations made by major professional societies are supported by high-quality evidence. Medical care that is not evidence-based can result in unnecessary care that burdens public finances, harms patients, and damages trust in the medical profession. Clearly, we must do a better job of figuring out the right treatments, for the right patients, at the right time. To meet this challenge, it is essential to improve our ability to capture reusable data at the point of care that can be used to improve care, discover new treatments, and make healthcare more efficient. To achieve this vision, we will need to shift financial incentives to reward data generation, change how we deliver care using AI, and continue improving the technological standards powering healthcare.
The Challenge and Opportunity of health data
Many have hailed health data collected during everyday healthcare interactions as the solution to some of these challenges. Congress directed the U.S. Food and Drug Administration (FDA) to increase the use of real-world data (RWD) for making decisions about medical products. However, FDA’s own records show that in the most recent year for which data are available, only two out of over one hundred new drugs and biologics approved by FDA were approved based primarily on real-world data.
A major problem is that our current model in healthcare doesn’t allow us to generate reusable data at the point of care. This is even more frustrating because providers face a high burden of documentation, and patients report repetitive questions from providers and questionnaires.
To expand a bit: while large amounts of data are generated at the point of care, these data lack the quality, standardization, and interoperability to enable downstream functions such as clinical trials, quality improvement, and other ways of generating more knowledge about how to improve outcomes.
By better harnessing the power of data, including results of care, we could finally build a learning healthcare system where outcomes drive continuous improvement and where healthcare value leads the way. There are, however, countless barriers to such a transition. To achieve this vision, we need to develop new strategies for the capture of high-quality data in clinical environments, while reducing the burden of data entry on patients and providers.
Efforts to achieve this vision follow a few basic principles:
- Data should be entered only once– by the person or entity most qualified to do so – and be used many times.
- Data capture should be efficient, so as to minimize the burden on those entering the data, allowing them to focus their time on doing what actually matters, like providing patient care.
- Data generated at the point of care needs to be accessible for appropriate secondary uses (quality improvement, trials, registries), while respecting patient autonomy and obtaining informed consent where required. Data should not be stuck in any one system but should flow freely between systems, enabling linkages across different data sources.
- Data need to be used to provide real value to patients and physicians. This is achieved by developing data visualizations, automated data summaries, and decision support (e.g. care recommendations, trial matching) that allow data users to spend less time searching for data and more time on analysis, problem solving, and patient care– and help them see the value in entering data in the first place.
Barriers to capturing high-quality data at the point of care:
- Incentives: Providers and health systems are paid for performing procedures or logging diagnoses. As a result, documentation is optimized for maximizing reimbursement, but not for maximizing the quality, completeness, and accuracy of data generated at the point of care.
- Workflows: Influenced by the prevailing incentives, clinical workflows are not currently optimized to enable data capture at the point of care. Patients are often asked the same questions at multiple stages, and providers document the care provided as part of free-text notes, which are frequently required for billing but can make it challenging to find information.
- Technology: Shaped by incentives and workflows, technology has evolved to capture information in formats that frequently lack standardization and interoperability.
Plan of Action
Plan of Action
Recommendation 1. Incentivize generation of reusable data at the point of care
Financial incentives are needed to drive the development of workflows and technology to capture high-quality data at the point of care. There are several payment programs already in existence that could provide a template for how these incentives could be structured.
For example, the Centers for Medicare and Medicaid Services (CMS) recently announced the Enhancing Oncology Model (EOM), a voluntary model for oncology providers caring for patients with common cancer types. As part of the EOM, providers are required to report certain data fields to CMS, including staging information and hormone receptor status for certain cancer types. These data fields are essential for clinical care, research, quality improvement, and ongoing care observation involving cancer patients. Yet, at present, these data are rarely recorded in a way that makes it easy to exchange and reuse this information. To reduce the burden of reporting this data, CMS has collaborated with the HHS Assistant Secretary for Technology Policy (ASTP) to develop and implement technological tools that can facilitate automated reporting of these data fields.
CMS also has a long-standing program that requires participation in evidence generation as a prerequisite for coverage, known as coverage with evidence development (CED). For example, hospitals that would like to provide Transcatheter Aortic Valve Replacement (TAVR) are required to participate in a registry that records data on these procedures.
To incentivize evidence generation as part of routine care, CMS should refine these programs and expand their use. This would involve strengthening collaborations across the federal government to develop technological tools for data capture, and increasing the number of payment models that require generation of data at the point of care. Ideally, these models should evolve to reward 1) high-quality chart preparation (assembly of structured data) 2) establishing diagnoses and development of a care plan, and 3) tracking outcomes. These payment policies are powerful tools because they incentivize the generation of reusable infrastructure that can be deployed for many purposes.
Recommendation 2. Improve workflows to capture evidence at the point of care
With the right payment models, providers can be incentivized to capture reusable data at the point of care. However, providers are already reporting being crushed by the burden of documentation and patients are frequently filling out multiple questionnaires with the same information. To usher in the era of the learning health system (a system that includes continuous data collection to improve service delivery), without increasing the burden on providers and patients, we need to redesign how care is provided. Specifically, we must focus on approaches that integrate generation of reusable data into the provision of routine clinical care.
While the advent of AI is an opportunity to do just that, current uses of AI have mainly focused on drafting documentation in free-text formats, essentially replacing human scribes. Instead, we need to figure out how we can use AI to improve the usability of the resulting data. While it is not feasible to capture all data in a structured format on all patients, a core set of data are needed to provide high-quality and safe care. At a minimum, those should be structured and part of a basic core data set across disease types and health maintenance scenarios.
In order to accomplish this, NIH and the Advanced Research Projects Agency for Health (ARPA-H) should fund learning laboratories that develop, pilot, and implement new approaches for data capture at the point of care. These centers would leverage advances in human-centered design and artificial intelligence (AI) to revolutionize care delivery models for different types of care settings, ranging from outpatient to acute care and intensive care settings. Ideally, these centers would be linked to existing federally funded research sites that could implement the new care and discovery processes in ongoing clinical investigations.
The federal government already spends billions of dollars on grants for clinical research- why not use some of that funding to make clinical research more efficient, and improve the experience of patients and physicians in the process?
Recommendation 3. Enable technology systems to improve data standardization and interoperability
Capturing high-quality data at the point of care is of limited utility if the data remains stuck within individual electronic health record (EHR) installations. Closed systems hinder innovation and prevent us from making the most of the amazing trove of health data.
We must create a vibrant ecosystem where health data can travel seamlessly between different systems, while maintaining patient safety and privacy. This will enable an ecosystem of health data applications to flourish. HHS has recently made progress by agreeing to a unified approach to health data exchange, but several gaps remain. To address these we must
- Increase standardization of data elements: The federal government requires certain data elements to be standardized for electronic export from the EHR. However, this list of data elements, called the United States Core Data for Interoperability (USCDI) currently does not include enough data elements for many uses of health data. HHS could rapidly expand the USCDI by working with federal partners and professional societies to determine which data elements are critical for national priorities, like vaccine safety and use, or protection from emerging pathogens.
- Enable writeback into the EHR: While current efforts focused on interoperability have focused on the ability to export EHR data, developing a vibrant ecosystem of health data applications that are available to patients, physicians, and other data users, requires the capability to write data back into the EHR. This would enable the development of a competitive ecosystem of applications that use health data generated in the EHR, much like the app store on our phones.
- Create widespread interoperability of data for multiple purposes: HHS has made great progress towards allowing health data to be exchanged between any two entities in our healthcare system, thanks to the Trusted Exchange Framework and Common Agreement (TEFCA). TEFCA could allow any two healthcare sites to exchange data, but unfortunately, participation remains spotty and TEFCA currently does not allow data exchange solely for research. HHS should work to close these gaps by allowing TEFCA to be used for research, and incentivizing participation in TEFCA, for example by making joining TEFCA a condition of participation in Medicare.
Conclusion
The treasure trove of health data generated during routine care has given us a huge opportunity to generate knowledge and improve health outcomes. These data should serve as a shared resource for clinical trials, registries, decision support, and outcome tracking to improve the quality of care. This is necessary for society to advance towards personalized medicine, where treatments are tailored to biology and patient preference. However, to make the most of these data, we must improve how we capture and exchange these data at the point of care.
Essential to this goal is evolving our current payment systems from rewarding documentation of complexity or time spent, to generation of data that supports learning and improvement. HHS should use its payment authorities to encourage data generation at the point of care and promote the tools that enable health data to flow seamlessly between systems, building on the success stories of existing programs like coverage with evidence development. To allow capture of this data without making the lives of providers and patients even more difficult, federal funding bodies need to invest in developing technologies and workflows that leverage AI to create usable data at the point of care. Finally, HHS must continue improving the standards that allow health data to travel seamlessly between systems. This is essential for creating a vibrant ecosystem of applications that leverage the benefits of AI to improve care.
This memo produced as part of the Federation of American Scientists and Good Science Project sprint. Find more ideas at Good Science Project x FAS
Confirming Hope: Validating Surrogate Endpoints to Support FDA Drug Approval Using an Inter-Agency Approach
To enable more timely access to new drugs and biologics, clinical trials are increasingly using surrogate markers in lieu of traditional clinical outcomes that directly measure how patients feel, function, or survive. Surrogate markers, such as imaging findings or laboratory measurements, are expected to predict clinical outcomes of interest. In comparison to clinical outcomes, surrogate markers offer an advantage in reducing the duration, size, and total cost of trials. Surrogate endpoints are considered to be “validated” if they have undergone extensive testing that confirms their ability to predict a clinical outcome. However, reviews of “validated” surrogate markers used as primary endpoints in trials supporting U.S. Food and Drug Administration (FDA) approvals suggest that many lack sufficient evidence of being associated with a clinical outcome.
Since 2018, FDA has regularly updated the publicly available “Table of Surrogate Endpoints That Were the Basis of Drug Approval or Licensure”, which includes over 200 surrogate markers that have been or would be accepted by the agency to support approval of a drug or biologic. Not included within the table is information regarding the strength of evidence for each surrogate marker and its association with a clinical outcome. As surrogate markers are increasingly being accepted by FDA to support approval of new drugs and biologics, it is imperative that patients and clinicians understand whether such novel endpoints are reflective of meaningful clinical benefits. Thus, FDA, in collaboration with other agencies, should take steps to increase transparency regarding the strength of evidence for surrogate endpoints used to support product approvals, routinely reassess the evidence behind such endpoints to continue justifying their use in regulatory decision-making, and sunset those that fail to show association with meaningful clinical outcomes. Such transparency would not only benefit the public, clinicians, and the payers responsible for coverage decisions, but also help shape the innovation landscape for drug developers to design clinical trials that assess endpoints truly reflective of clinical efficacy.
Challenge and Opportunity
To receive regulatory approval by FDA, new therapeutics are generally required to be supported by “substantial evidence of effectiveness” from two or more “adequate and well-controlled” pivotal trials. However, FDA has maintained a flexible interpretation of this guidance to enable timely access to new treatments. New drugs and biologics can be approved for specific disease indications based on pivotal trials measuring clinical outcomes (how patients feel, function, or survive). They can also be approved based on pivotal trials measuring surrogate markers that are meant to be proxy measures and expected to predict clinical outcomes. Examples of such endpoints include changes in tumor size as seen on imaging or blood laboratory tests such as cholesterol.
Surrogate markers are considered “validated” when sufficient evidence demonstrates that the endpoint reliably predicts clinical benefit. Such validated surrogate markers are typically the basis of traditional FDA therapeutics approval. However, FDA has also accepted the use of “unvalidated” surrogate endpoints that are reasonably likely to predict clinical benefit as the basis of approval of new therapeutics, particularly if they are being used to treat or prevent a serious or life-threatening disease. Under expedited review pathways, such as accelerated approval that grant drug manufacturers faster FDA market authorization using unvalidated surrogate markers, manufacturers are required to complete an additional clinical trial after approval to confirm the predicted clinical benefit. Should the manufacturer fail to do so, FDA has the authority to withdraw that drug’s particular indication approval.
For drug developers, the use of surrogate markers in clinical trials can shorten the duration, size, and total cost of the pivotal trial. Over time, FDA has increasingly allowed for surrogate markers to be used as primary endpoints in pivotal trials, allowing for shorter clinical trial testing periods and thus faster market access. Moreover, use of unvalidated surrogate markers has grown outside of expedited review pathways such as accelerated approval. One analysis of FDA approved drugs and biologics that received “breakthrough therapy designation” found that among those that received traditional approval, over half were based on pivotal trials using surrogate markers.
While basing FDA approval on surrogate markers can enable more timely market access to novel therapeutics, such endpoints also involve certain trade-offs, including the risk of making erroneous inferences and diminishing certainty about the medical product’s long-term clinical effect. In oncology, evidence suggests that most validation studies of surrogate markers find low correlations with meaningful clinical outcomes such as overall survival or a patient’s quality of life. For instance, in a review of 15 surrogate validation studies conducted by the FDA for oncologic drugs, only one was found to demonstrate a strong correlation between surrogate markers and overall survival. Another study suggested that there are weak or missing correlations between surrogate markers for solid tumors and overall survival. A more recent evaluation found that most surrogate markers used as primary endpoints in clinical trials to support FDA approval of drugs treating non-oncologic chronic disease lack high-strength evidence of associations with clinical outcomes.
Section 3011 of the 21st Century Cures Act of 2016 amended the Federal Food, Drug, and Cosmetic Act to mandate FDA publish a list of “surrogate endpoints which were the basis of approval or licensure (as applicable) of a drug or biological product” under both accelerated and traditional approval pathways. While FDA has posted surrogate endpoint tables for adult and pediatric disease indications that fulfil this legislative requirement, missing within these tables is any justification for surrogate selection, including evidence supporting validation. Without this information, patients, prescribers, and payers are left uncertain about the actual clinical benefit of therapeutics approved by the FDA based on surrogate markers. Instead, drug developers have continued to use this table as a guide in designing their clinical trials, viewing the included surrogate markers as “accepted” by the FDA regardless of the evidence (or lack thereof) undergirding them.
Plan of Action
Recommendation 1. FDA should make more transparent the strength of evidence of surrogate markers included within the “Adult Surrogate Endpoint Table” as well as the “Pediatric Surrogate Endpoint Table.”
Previously, agency officials stated that the use of surrogate markers to support traditional approvals was usually based, at a minimum, on evidence from meta-analyses of clinical trials demonstrating an association between surrogate markers and clinical outcomes for validation. However, more recently, FDA officials have indicated that they consider a “range of sources, including mechanistic evidence that the [surrogate marker] is on the causal pathway of disease, nonclinical models, epidemiologic data, and clinical trial data, including data from the FDA’s own analyses of patient- and trial-level data to determine the quantitative association between the effect of treatment on the [surrogate marker] and the clinical outcomes.” Nevertheless, what specific evidence and how the agency weighed such evidence is not included as part of their published tables of surrogate endpoints, leaving unclear to drug developers as well as patients, clinicians, and payers the strength of the evidence behind such endpoints. Thus, this serves as an opportunity for the agency to enhance their transparency and communication with the public.
FDA should issue a guidance document detailing their current thinking about how surrogate markers should be validated and evaluated on an ongoing basis. Within the guidance, the agency could detail the types of evidence that would be considered to establish surrogacy.
FDA should also include within the tables of surrogate endpoints, a summary of evidence for each surrogate marker listed. This would provide justification (through citations to relevant articles or internal analyses) so that all stakeholders understand the evidence establishing surrogacy. Moreover, FDA can clearly indicate within the tables which clinical outcomes each surrogate marker listed is thought to predict.
FDA should also publicly report on an annual basis a list of therapeutics approved by the agency based on clinical trials using surrogate markers as primary endpoints. This coupled with the additional information around strength of evidence for each surrogate marker would allow patients and clinicians to make more informed decisions around treatments where there may be uncertainty of the therapeutic’s clinical benefit at the time of FDA approval.
Recently, FDA’s Oncology Center for Excellent through Project Confirm has made additional efforts to communicate that status of required postmarketing studies meant to confirm clinical benefit of drugs for oncologic disease indications that received accelerated approval. FDA could further expand this across therapeutic areas and approval pathways by publishing a list of ongoing postmarketing studies for therapeutics where approval was based on surrogate markers that are intended to confirm clinical benefit.
FDA should also regularly convene advisory committees to allow for independent experts to review and vote on recommendations around the use of new surrogate markers for disease indications. Additionally, FDA should regularly convene these advisory committees to re-evaluate the use of surrogate markers based on current evidence, especially those not supported by high-strength evidence demonstrating their association with clinical outcomes. At a minimum, FDA should convene such advisory committees focused on re-examining surrogate markers listed on their publicly available tables annually. In 2024, FDA convened the Oncologic Drugs Advisory Committee to discuss the use of the surrogate marker, minimal residual disease as an endpoint for multiple myeloma. Further such meetings including for those “unvalidated” endpoints would provide FDA opportunity to re-examine their use in regulatory decision-making.
Recommendation 2. In collaboration with the FDA, other federal research agencies should contribute evidence generation to determine whether surrogate markers are appropriate for use in regulatory decision-making, including approval of new therapeutic products and indications for use.
Drug manufacturers that receive FDA approval for products based on unvalidated surrogate markers may not be incentivized to conduct studies that demonstrate a lack of association between such surrogate markers with clinical outcomes. To address this, the Department of Health and Human Services (HHS) should establish an interagency working group including FDA, National Institutes of Health (NIH), Patient Centered Outcomes Research Institute (PCORI), Advanced Research Projects Agency for Health (ARPA-H), Centers for Medicare and Medicaid Services (CMS) and other agencies engaged in biomedical and health services research. These agencies could collaboratively conduct or commission meta-analyses of existing clinical trials to determine whether there is sufficient evidence to establish surrogacy. Such publicly-funded studies would then be brought to FDA advisory committees to be considered by members in making recommendations around the validity of various surrogate endpoints or whether any endpoints without sufficient evidence should be sunset. NIH in particular should prioritize funding large-scale trials aimed at validating important surrogate outcomes.
Through regular collaboration and convening, FDA can help guide the direction of resources towards investigating surrogate markers of key regulatory as well as patient, clinician, and payer interest to strengthen the science behind novel therapeutics. Such information would also be invaluable to drug developers in identifying evidence-based endpoints as part of their clinical trial design, thus contributing to a more efficient research and development landscape.
Recommendation 3. Congress should build upon the provisions related to surrogate markers that passed as part of the 21st Century Cures Act of 2016 in their “Cures 2.0” efforts.
The aforementioned interagency working group convened by HHS could be authorized explicitly through legislation coupled with funding specifically for surrogate marker validation studies. Congress should also mandate that FDA and other federal health agencies re-evaluate listed surrogate endpoints on an annual basis with additional reporting requirements. Additionally, through legislation, FDA could also be granted explicit authority for those endpoints where there is no clear evidence of their surrogacy to sunset them, thus preventing future drug candidates from establishing efficacy based on flawed endpoints. Congress should also require routine reporting from FDA on the status of the interagency working group focused on surrogate endpoints as well as other metrics including a list of new therapeutic approvals based on surrogate markers, expansion of the existing surrogate marker tables on FDA’s website to include the evidence of their surrogacy, and issuance of a guidance document detailing what scientific evidence would be considered by the agency in validating and re-evaluating surrogate markers.
Conclusion
FDA increasingly has allowed new drugs and biologics to be approved based on surrogate markers that are meant to be predictive of meaningful clinical outcomes demonstrating that patients feel better, function better, and survive longer. Although the agency has made more clear what surrogate endpoints could be or are being used to support approval, significant gaps exist in the evidence demonstrating that these novel endpoints are associated with meaningful clinical outcomes. Continued use of surrogate endpoints with little association with clinical benefit leaves patients and clinicians without assurance that novel therapeutics approved by FDA are meaningfully effective, as well as payers responsible for coverage decisions. Transparency of the evidence supporting clinical endpoints is urgently needed to mitigate this uncertainty around new drug approvals, including for drug developers as they continue clinical trials for therapeutic candidates seeking FDA approval. FDA should and in collaboration with other federal biomedical research agencies, routinely re-evaluate surrogate endpoints to determine their continued use in therapeutic innovation. Such regular re-evaluation that informs regulatory decision-making will strengthen FDA’s credibility and ensure accountability of the agency, tasked with ensuring the safety and efficacy of drugs and other medical products as well as with shaping the innovation landscape.
This memo produced as part of the Federation of American Scientists and Good Science Project sprint. Find more ideas at Good Science Project x FAS
Yes. In 2016, eteplirsen (Exondys 51) was granted accelerated approval for the treatment of Duchenne muscular dystrophy (DMD) against the recommendation of an advisory committee and FDA’s own scientific staff. Concerns were raised that the approval was based on a small clinical trial that showed that eteplirsen led to a small increase in protein dystrophin, a surrogate marker. Three additional approvals for similar DMD drugs have been made based on the same surrogate endpoint. However, no studies have been completed providing confirmation of clinical benefit.
In 2021, aducanumab (Aduhelm) was granted accelerated approval for the treatment of Alzheimer’s disease against the recommendation of an advisory committee and FDA’s scientific staff. Concerns were raised that the approval was based on a surrogate marker, beta-amyloid levels, which has not been found to correlate with cognitive or function changes for Alzheimer’s disease patients. In particular, FDA’s internal statistical review team found no association between changes to the surrogate marker and the clinical outcomes reported in pivotal trials.
Industry may claim that such re-evaluation and potential removal of continued unvalidated surrogate endpoints would slow down the pace of innovation and thus, patient access to novel therapeutics. However, it is more likely this would instead enable more efficient drug development in providing manufacturers, particularly smaller companies with surrogate endpoints that not only decrease the duration and cost of clinical trials, but that also have strong evidence of association with meaningful clinical outcomes. This may also mitigate the need for postmarketing requirements for manufacturers meant to confirm clinical benefit if adequate validation is conducted through FDA and other federal agencies.
No. Having FDA in collaboration with other federal health agencies to validate surrogate endpoints would not halt the use of unvalidated surrogate endpoints reasonably likely to predict clinical benefit. Expedited regulatory pathways such as accelerated approval that are codified by law allowing manufacturers to use unvalidated surrogate markers as endpoints in pivotal clinical trials will still be available for manufacturers. Instead, this creates a process for re-evaluation such that unvalidated surrogate endpoints are not forever left unvalidated, but instead examined within a timely manner to inform their continued use in supporting FDA approval. Ultimately, patients and clinicians want drugs that meaningfully work to treat or prevent against a disease or condition. Routine re-evaluation and validation of surrogate endpoints would provide assurance that for those therapeutics whose approval is based off of these novel endpoints that the FDA approved treatment is clinically effective.
FDA’s function as a regulator is to evaluate the evidence that is brought before them by industry sponsors. To do so effectively, the evidence must be available. This is often not the case, particularly for new surrogate markers as there may not be commercial incentive to do so, particularly if after approval, a surrogate endpoint is found to be not associated with a meaningful clinical outcome. Thus, the involvement of multiple federal biomedical research agencies including NIH and ARPA-H alongside FDA can play an instrumental role in conducting or funding studies demonstrating a clear association between surrogate marker and clinical outcome. Already, several institutes within the NIH are engaged in biomarker development and in supporting validation. Collaboration between NIH institutes with expertise as well as other agencies engaged in translational research with FDA will enable validation of surrogate markers to inform regulatory decision-making of novel therapeutics.
Under the Prescription Drug User Fee Act VII passed in 2022, FDA was authorized to establish the Rare Disease Endpoint Advancement (RDEA) pilot program. This program is intended to foster the development of novel endpoints for rare diseases through FDA collaboration with industry sponsors. with proposed novel endpoints for a drug candidate, opportunities for stakeholders including the public to inform such endpoint development, and greater FDA staff capacity to help develop novel endpoints for rare diseases. Such a pilot program could be further expanded to not only develop novel endpoints, but to also develop approaches for validating novel endpoints such as surrogate markers and communicating the strength of evidence to the public.
Payers such as Medicare have also taken steps to enable postmarket evidence generation including for drugs approved by FDA based on surrogate endpoints. Following the accelerated approval of aducanumab (Aduhelm), the Centers for Medicare and Medicaid Services (CMS) issued a national coverage determination under the coverage with evidence development (CED) program, conditioning coverage of this class of drugs to studies approved by CMS approved by FDA based on a surrogate endpoint with access only available through randomized controlled trials assessing meaningful clinical outcomes. Further evaluation of surrogate endpoints informing FDA approval can be beneficial for payers as they make coverage decisions. Additionally, coverage and reimbursement could also be tied to evidence for such surrogate endpoints, providing additional incentive to complete and communicate the findings from such studies.
A Cross-Health and Human Services Initiative to Cut Wasteful Spending and Improve Patient Lives
Challenge and Opportunity
Many common medical practices do not have strong evidence behind them. In 2019, a group of prominent medical researchers—including Robert Califf, the former Food and Drug Administration (FDA) Commissioner—undertook the tedious task of looking into the level of evidence behind 2,930 recommendations in guidelines issued by the American Heart Association and the American College of Cardiology. They asked one simple question: how many recommendations were supported by multiple small randomized trials or at least one large trial? The answer: 8.5%. The rest were supported by only one small trial, by observational evidence, or just by “expert opinion only.”
For infectious diseases, a team of researchers looked at 1,042 recommendations in guidelines issued by the Infectious Diseases Society of America. They found that only 9.3% were supported by strong evidence. For 57% of the recommendations, the quality of evidence was “low” or “very low.” And to make matters worse, more than half of the recommendations considered low in quality of evidence were still issued as “strong” recommendations.
In oncology, a review of 1,023 recommendations from the National Comprehensive Cancer Network found that “…only 6% of the recommendations … are based on high-level evidence”, suggesting “a huge opportunity for research to fill the knowledge gap and further improve the scientific validity of the guidelines.”
Even worse, there are many cases where not only is a common medical treatment lacking the evidence to support it, but also one or more randomized trials have shown that the treatment is useless or even harmful! One of the most notorious examples is that of the anti-arrhythmic drugs given to millions of cardiac patients in the 1980s. Cardiologists at the time had the perfectly logical belief that since arrhythmia (irregular heartbeat) leads to heart attacks and death, drugs that prevented arrhythmia would obviously prevent heart attacks and death. In 1987, the National Institutes of Health (NIH) funded the Cardiac Arrhythmia Suppression Trial (CAST) to test three such drugs. One of the drugs had to be pulled after just a few weeks, because 17 patients had already died compared with only three in the placebo group. The other two drugs similarly turned out to be harmful, although it took several months to see that patients given those drugs were more than two times as likely to die. According to one JAMA article, “…there are estimates that 20,000 to 75,000 lives were lost each year in the 1980s in the United States alone…” due to these drugs. The CAST trial is a poignant reminder that doctors can be convinced they are doing the best for their patients, but they can be completely wrong if there is not strong evidence from randomized trials.
In 2016, randomized trials of back fusion surgery found that it does not work. But a recent analysis by the Lown Institute found that the Centers for Medicare & Medicaid Services (CMS) spent approximately $2 billion in the past 3 years on more than 200,000 of these surgeries.
There are hundreds of additional examples where medical practice was ultimately proven wrong. Given how few medical practices, even now, are actually supported by strong evidence, there are likely many more examples of treatments that either do not work or actively cause harm. This is not only wasted spending, but also puts patients at risk.
We can do better – both for patients and for the federal budget – if we reduce the use of medical practices that simply do not work.
Plan of Action
The Secretary of Health and Human Services should create a cross-division committee to develop an extensive and prioritized list of medical practices, products, and treatments that need evidence of effectiveness, and then roll out an ambitious agenda to run randomized clinical trials for the highest-impact medical issues.
That is, the CMS needs to work with the NIH and the FDA, and the Centers for Disease Control and Prevention (CDC) to develop a prioritized list of medical treatments, procedures, drugs, and devices with little evidence behind them and for which annual spending is large and the health impacts could be most harmful. Simultaneously, the FDA needs to work with its partner agencies to identify drugs, vaccines, and devices with widespread medical usage that need rigorous post-market evaluation. This includes drugs with off-label uses, oncology regimens that have never been tested against each other, surrogate outcomes that have not been validated against long-term outcomes, accelerated approvals without the needed follow-up studies, and more.
With priority lists available, the NIH could immediately launch trials to evaluate the effectiveness of the identified treatments and practices to ensure effective health and safety. The Department should report to Congress on a yearly basis as to the number and nature of clinical trials in progress, and eventually the results of those trials (which should also be made available on a public dashboard, with any resulting savings). The project should be ongoing for the indefinite future, and over time, HHS should explore ways to have artificial intelligence tools identify the key unstudied medical questions that deserve a high-value clinical trial.
Expected opponents to any such effort will be pharmaceutical, biotechnology and device companies and their affiliated trade associations, whose products might come under further scrutiny, and professional medical associations who are firmly convinced that their practices should not be questioned. Their lobbying power might be considerable, but the intellectual case behind the need for rigorous and unbiased studies is unquestionable, particularly when billions of federal dollars and millions of patients’ lives and health are at stake.
Conclusion
Far too many medical practices and treatments have not been subjected to rigorous randomized trials, and the divisions of Health and Human Services should come together to fix this problem. Doing so will likely lead to billions of dollars in savings and huge improvements to patient health.
This memo produced as part of the Federation of American Scientists and Good Science Project sprint. Find more ideas at Good Science Project x FAS
Supporting Device Reprocessing to Reduce Waste in Health Care
The U.S. healthcare system produces 5 million tons of waste annually, or approximately 29 pounds per hospital bed daily. Roughly 80 percent of the healthcare industry’s carbon footprint comes from the production, transportation, use, and disposal of single-use devices (SUDs), which are pervasive in the hospital. Notably, 95% of the environmental impact of single-use medical products results from the production of those products.
While the Food and Drug Administration (FDA) oversees new devices being brought to market, it is up to the manufacturer to determine whether a device will be marketed as single-use or multiple-use. Manufacturers have a financial incentive to market devices for “single-use” or “disposable” as marketing a device as reusable requires expensive cleaning validations.
In order to decrease healthcare waste and environmental impact, FDA leads on identifying reusable devices that can be safely reprocessed and incentivizing manufacturers to test the reprocessing of their device. This will require the FDA to strengthen its management of single-use and reusable device labeling. Further, the Veterans Health Administration, the nation’s largest healthcare system, should reverse the prohibition on reprocessed SUDs and become a national leader in the reprocessing of medical devices.
Challenge and Opportunity
While healthcare institutions are embracing decarbonization and waste reduction plans, they cannot do this effectively without addressing the enormous impact of single-use devices (SUDs). The majority of research literature concludes that SUDs are associated with higher levels of environmental impact than reusable products.
FDA regulations governing SUD reprocessing make it extremely challenging for hospitals to reprocess low-risk SUDs, which is inconsistent with the FDA’s “least burdensome provisions.” The FDA requires hospitals or commercial SUD reprocessing facilities to act as the device’s manufacturer, meaning they must follow the FDA’s rules for medical device manufacturers’ requirements and take on the associated liabilities. Hospitals are not keen to take on the liability of a manufacturer, yet commercial reprocessors do not offer many lower-risk devices that can be reprocessed.
As a result, hospitals and clinics are no longer willing to sterilize SUDs through methods like autoclaving even despite documentation showing that sterilization is safe and precedent showing that similar devices have been safely sterilized and reused for many years without adverse events. Many devices, including pessaries for pelvic organ prolapse and titanium phacoemulsification tips for cataract surgery, can be safely reprocessed in their clinical use. These products, given their risk profile, need not be subject to the FDA’s full medical device manufacturer requirements.
Further, manufacturers are incentivized to bring SUDs to market quicker than those that may be reprocessed. Manufacturers often market devices as single-use solely because the original manufacturer chose not to conduct expensive cleaning and sterilization validations, not because such cleaning and sterilization validations cannot be done. FDA regulations that govern SUDs should be better tailored to each device so that clinicians on the frontlines can provide appropriate and environmentally sustainable health care.
Reprocessed devices cost 25 to 40% less. Thus, the use of reprocessed SUDs can reduce costs in hospitals significantly — about $465 million in 2023. Per the Association of Medical Device Reprocessors, if the reprocessing practices of the top 10% performing hospitals were maximized across all hospitals that use reprocessed devices, U.S. hospitals could have saved an additional $2.28 billion that same year. Indeed, enabling and encouraging the use of reprocessed SUDs can also yield significant cost reductions without compromising patient care.
Plan of Action
As the FDA began regulating SUD reprocessing in 2000, it is imperative that the FDA take the lead on creating a clear, streamlined process for clearing or approving reusable devices in order to ensure the safety and efficacy of reprocessed devices. These recommendations would permit healthcare systems to reprocess and reuse medical devices without fear of noncompliance by the Joint Commission or Centers for Medicare and Medicaid Services that reply on FDA regulations. Further, the nation’s largest healthcare system, the Veterans Health Administration, should become a leader in medical device reprocessing, and lead on showcasing the standard of practice for sustainable healthcare.
- FDA should publish a list of SUDs that have a proven track record of safe reprocessing to empower hospitals to reduce waste, costs, and environmental impact without compromising patient safety. The FDA should change the labels of single-use devices to multi-use when reuse by hospitals is possible and validated via clinical studies, as the “single-use” label has promoted the mistaken belief that SUDs cannot be safely reprocessed. Per the FDA, the single-use label simply means a given device has not undergone the original equipment manufacturer (OEM) validation tests necessary to label a device “reusable.” The label does not mean the device cannot be cleared for reprocessing.
- In order to help governments and healthcare systems prioritize the environmental and cost benefits of reusable devices over SUDs, FDA should incentivize applications of reusable or commercially reprocessable devices, such as through expediting review. The FDA can also incentivize use of reprocessed devices through payments to hospitals for meeting reprocessing benchmarks.
- The FDA should not subject low-risk devices that can be safely reprocessed for clinical use to full device manufacturer requirements. The FDA should further encourage healthcare procurement staff by creating an accessible database of devices cleared for reprocessing and alerting healthcare systems about regulated reprocessing options. In doing so, the FDA can help reduce the burden on hospitals in reprocessing low-risk SUDs and encourage healthcare systems to sterilize SUDs through methods like autoclaving.
- As the only major health system in the U.S. to prohibit the use of reprocessed SUDs, the U.S. Veterans Health Administration should reverse its prohibition as soon as possible. This prohibition likely remains because of outdated determinations of risks, which comes at major costs for the environment and Americans. Doing so would be consistent with the FDA’s conclusions that reprocessed SUDs are safe and effective.
- FDA should recommend that manufacturers publicly report the materials used in the composition of devices so that end-users can more easily compare products and determine the environmental impact of devices. As explained by AMDR, some Original Equipment Manufacturer (OEM) practices discourage or fully prevent the use of reprocessed devices. It is imperative that the FDA vigorously track and impede these practices. Not only will requiring public reporting device composition help healthcare buyers make more informed decisions, it will also help promote a more circular economy that uplifts sustainability efforts.
Conclusion
To decrease costs, waste, and environmental impact, the healthcare sector urgently needs to increase its use of reusable devices. One of the largest barriers is FDA requirements that result in needlessly stringent requirements of hospitals, hindering the adoption of less wasteful, less costly reprocessed devices.
FDA’s critical role in medical device labeling, clearing, or approving more devices as reusable, has down market implications and influences many other regulatory and oversight bodies, including the Centers for Medicare & Medicaid Services (CMS), the Association for the Advancement of Medical Instrumentation (AAMI), the Joint Commission, hospitals, health care offices, and health care providers. It is essential for the FDA to step up and take the lead in revising the device reprocessing pipeline.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
A Peer Support Service Integrated Into the 988 Lifeline
A peer support option should be integrated into the 988 Suicide and Crisis Lifeline so that 988 service users can choose to connect with specialists based on a shared lived experience. As people and communities become more siloed, the risk of mortality and morbidity increases. Social connectedness is a critical protective factor. A peer support service would allow individuals to receive support built on a lived experience that is common to both the service user and the specialist. It should be free and easily accessible through phone call and text messaging. This service is especially timely, following the 2020 rollout of the 988 Suicide and Crisis Lifeline, as well as recent peer support initiatives at the federal and state levels.
While the efficacy of peer support is most known with mental illness, it has successfully helped a range of individuals including cancer patients, people experiencing homelessness, racial minorities, veterans, and formerly incarcerated people. The peer support service should provide support for all kinds of lived experience, including experiences with: disability resulting from poor physical or mental health, substance use, suicidal ideation, veteran-connected disability, financial insecurity, homelessness, domestic violence and family court, nonnuclear family structures or living alone, former incarceration, and belonging to racial or ethnic minority groups. The service should be a preventative intervention as well as complimentary assistance for those in recovery or treatment.
Challenge and Opportunity
The United States faces an “epidemic of loneliness and isolation.” While individuals from all backgrounds are afflicted, the most vulnerable and underserved members of society have suffered the most. A range of challenging circumstances cause this suffering, including poor physical or mental health, nontraditional living conditions, and historic inequality. Not having anyone who shares one’s lived experience is isolating. These challenging life circumstances take an emotional toll, and they become risk factors for a slew of physical and mental health conditions that increase morbidity and decrease life expectancy.
In addition to the social costs, these outcomes have an increasingly devastating economic impact. Health care costs are projected to account for 20% of the U.S. economy by 2031. Psychiatric hospitalizations are steadily increasing, and a shortage of psychiatric inpatient beds is rampant.
Social connectedness alleviates physical and mental burdens. Peer support offers a cost-effective intervention for prevention and recovery. It reduces spending on both physical and mental illnesses, and it reduces psychiatric hospitalizations, saving an average of $4.76 for every $1 spent on peer support. In a New York City-based peer support program, service users saved about $2,000 per month in Medicaid costs and had 2.9 fewer hospitalizations per year.
Telephone-based peer support has life-saving outcomes, too. Over telephone, peer support led to a 15% increase in women’s mammography screening rates, with the highest increase among women of low-to-middle income. Telephone-based peer support increased breastfeeding rates by 14% and reduced breastfeeding dissatisfaction by 10% among first-time mothers, and it led to a 10% change in diet among patients with heart disease. While peer support would not solve national crises of homelessness or rising healthcare costs, it would ease them by fostering community empowerment and self-reliance and reducing federal intervention.
In 2023, SAMHSA rolled out “National Model Standards for Peer Support Certification”. This guide provides recommendations for how each state can integrate its own “peer mental health workforce across all elements of the healthcare system.” In its current form, SAMHSA’s strategy targets lived experiences with substance use and mental health. A broader scope would assist and empower more underserved members of the community. Following the momentum of SAMHSA’s initiative, now is the most optimal time to integrate a peer support service into the 988 Lifeline.
Peer support exists in the U.S., but services are spread thin across private and nonprofit sectors. The Restoring Hope for Mental Health and Well-Being Act of 2022 authorized $1.7 trillion in funding until FY2027 for various health initiatives, including peer support mental health services. This funding relies on organizations and institutions to independently implement peer support services based on community needs. However, a fragmented system can result in underuse, limited accessibility, and a varied quality of service, with pockets of the United States lacking any service at all. It also poses privacy concerns about how individuals’ data are stored and used, as well as cybersecurity vulnerabilities with smaller organizations that may lack a robust security infrastructure.
A peer support service that is integrated into the 988 Lifeline would ensure that all Americans have equal access to a high-quality, confidential peer support network. The standardization of the 988 Lifeline is a prime example of successful implementation. Its transition from a 10-digit number to a three-digit dial led to a 33% average increase in in-state call volume over four months. Standardization shifted funding from primarily private and nonprofit initiatives and donations to stable public sector support. As a result, call pickup rates rose from 70% to 93%, and wait times dropped from 2 minutes 20 seconds to just 35 seconds.
The 988 Lifeline also adheres to privacy and confidentiality protocols in line with the Health Insurance Portability and Accountability Act (HIPAA) Security Rule. Under these protocols, the 988 Lifeline retains minimal information about callers and texters, and this information stays private and securely stored. A peer support service with similar safeguards would help both service users and peer support workers (PSWs) feel safe when sharing sensitive information.
Peer support for mental health has gained traction recently. The National Alliance for Mental Illness (NAMI) offers peer-to-peer courses, although these are currently limited in their duration and available locations. Last year, Congress expressed an interest in peer support mental health services: a “Supporting All Students Act” was proposed in the Senate for “peer and school-based mental health support.” This endeavor especially targets the escalating suicide crisis among youths in the form of a peer-to-peer suicide prevention. The Wisconsin Department of Public Instruction also recently introduced Peer-to-Peer Suicide Prevention Grants. The federal and state-level governments clearly recognize the value of peer support work. Still, they underutilize its potential.
Loneliness and isolation are at an all-time high in the United States. However, they stem from a multitude of causes. An empathic connection with a peer who has lived the same experience has immense potential for healing and recovery in both the PSW and the service user. The 988 Lifeline should include an integrated peer support service for both mental health and a broad range of lived experiences. While there exist some peer support services for conditions not related to mental health, these are not standardized or easily accessible to the entire American population. Many peer-operated warmlines (i.e., support lines) exist. However, they are spread across dozens of phone numbers and websites with varied and limited sources of funding. A single peer support service integrated into the 988 Lifeline would make peer support universally available. This service would address the emotional toll that accompanies a wide range of challenging life circumstances.
In short, a peer support service integrated into the 988 Lifeline would make the efforts of existing peer support organizations even more effective as it would:
- Standardize training, quality of care, and confidentiality,
- Expand the kinds of lived experience that are available for peer support, and
- Maximize the distribution of high-quality services across the U.S., with a focus on reaching high-risk populations.
Action Plan
The peer support service should begin by covering only a few kinds of lived experiences. After this implementation, the service should be expanded to cover a broader range of experiences.
Recommendation 1. Create a Peer Support Task Force (PSTF).
A PSTF should be established within SAMHSA. The secretary of the U.S. Department of Health and Human Services (HHS) should work with SAMHSA’s assistant secretary for mental health and substance use to establish this temporary task force.
The PSTF should lead the implementation of the peer support service, acting as an interagency task force that coordinates with partners across the public, private, and nonprofit sectors. This partnership will ensure that different lived experiences are accounted for and that existing resources are used effectively. The PSTF should collaborate with federal agencies, including the Department of Veterans Affairs (VA) and the Indian Health Service (IHS), as well as with advocacy organizations like NAMI and the American Cancer Society that champion the needs of people with specific lived experiences.
The PSTF should be charged with the following recommendations to start.
Recommendation 2. Integrate a peer support option into the 988 Lifeline.
Under the authority of the PSTF, integrating a peer support option into the 988 Lifeline could bypass additional Congressional action.
In the first pass, the integrated peer support option should cover only a few kinds of lived experiences (e.g., suicide and behavioral crises, veteran-connected disability).
- Before scaling the peer support service across the United States, the PSTF should implement a pilot program to test and refine protocols. The PSTF should select 988 Lifeline centers in states that already have a strong peer support program, and it should pilot peer support call and text services. The pilot program should select a few peer support needs to test first (e.g., suicide and behavioral crises, veteran-connected disability), as a narrow scope will be easier to assess for the first pass.
- The PSTF should then incorporate feedback from this pilot program as it scales up and integrates a peer support option into the nationwide 988 Lifeline.
- The PSTF should coordinate with the Federal Communications Commission (FCC) to add a caller menu option for peer support into the 988 Lifeline. (E.g., “Press 4 for peer support.”)
- The PSTF should coordinate with the FCC and U.S. Digital Service to implement a telephone triage, so the service user can submit a request for a specific kind of peer support and then be routed to a PSW on shift who has a lived experience that best matches the submitted request.
- The PSTF should facilitate the 988 Lifeline’s partnership with existing local and national peer support organizations and warmlines. Partnering with these existing organizations would bolster the 988 Lifeline’s capacity to provide peer support. Furthermore, states that already have a strong peer support certification in place could easily integrate their trained PSWs into the 988 Lifeline services using their existing infrastructure and expertise.
- The PSTF should coordinate with the FCC to incorporate a peer support text messaging option within the 988 Lifeline’s text and chat services. (For example, service users could text “PEER” to 9-8-8 and be routed to a PSW on shift.)
- The PSTF should implement a public campaign to the general public clarifying that the 988 Lifeline will remain as a suicide and crisis hotline, but it will also provide access to broader peer support services. The available peer support services should be clearly outlined on the 988 Lifeline website.
PSWs should work in call centers alongside 988 Lifeline phone and text specialists.
Recommendation 3. Develop an action plan to fund and sustain the integration of the peer support service.
The peer support service would need funding to become integrated into the 988 Lifeline. The government has recently approved funds for mental health initiatives, such as with the Restoring Hope for Mental Health and Well-Being Act of 2022. In 2023, HHS announced an additional $200 million in funding for the 988 Lifeline, but overall, HHS has granted almost $1.5 billion in total toward the 988 Lifeline. Similar avenues of funding should help jumpstart the integration of the peer support service.
For continued maintenance, fees for the peer support service should apply in a similar fashion to 9-1-1. That is, service users should not pay each time they access the 988 Lifeline or its integrated peer support service. Some U.S. states have already passed 9-8-8 implementation legislation that allows a monthly flat fee to be collected through telephone and wireless service providers, as permitted in the “National Suicide Hotline Designation Act of 2020”. The remaining states should be encouraged to pass similar legislation. The fee should remain in an account that is spent only for the maintenance of the 988 Lifeline and peer support services. If a fee is collected, then the FCC should provide an annual report on these fees and their usage.
Funding for the peer support service’s integration into the 988 Lifeline would entail the following:
- Maintenance of the peer support service
- Financial compensation for trainers who will train and mentor the PSWs
- Recruitment of trainers and PSWs
- The development of training resources for PSWs
Recommendation 4. Establish state-level standardized peer support training and certification.
PSWs should learn skills that are commonly taught to 988 Lifeline specialists, including active listening and recovery-oriented language. They should learn how to share their own stories, navigate challenging conversations, maintain boundaries, and self-care. Their training should be standardized at the state level, and it should be based on the “National Model Standards for Peer Support Certification” established by SAMHSA.
While most U.S. states have some version of peer support certification for mental health and/or substance use recovery, the PSTF should work with advocacy organizations to ensure that the state-level standardized training accounts for the peer support needs and demographics of each state. The peer support training should address the needs of people with a diverse range of lived experiences. Peer support needs can also be studied using caller outcome data that are recorded by 988 Lifeline specialists.
The PSTF should recruit trainers who will train and mentor the PSWs. To start, the PSTF should liaise within SAMHSA and with existing peer support organizations to coordinate this recruitment.
Recommendation 5. Establish a nation-wide system for the recruitment and training of PSWs.
Recruit individuals who would like to use their own lived experience to help other community members. Recruitment should occur through hospitals, clinics, as well as peer-run and community-based organizations to maximize recruitment pathways.
Current 988 Lifeline phone and text specialists could also be good candidates for the peer support service. Many are motivated by their own lived experiences and are already trained to handle calls across multiple helplines. 988 Lifeline specialists are instructed to focus on the service user seeking help and not share about themselves. However, specialists’ own lived experiences––if they are comfortable sharing them––represent untapped potential.
Once recruited, individuals who are training to become PSWs should attend live training sessions online with a peer cohort.
Recommendation 6. Expand the peer support service to cover a diverse range of lived experiences.
After successfully establishing the peer support service, it should be expanded upon to provide support for a broad and diverse range of lived experiences. The available peer support services should continue to be updated and outlined on the 988 Lifeline website.
Conclusion
A peer support service should be integrated into the 988 Lifeline. A service that caters to all kinds of lived experience paves way for a more empowered and self-reliant community. It fosters a societal mindset of helping each other based on shared lived experience in an empathic and healthy way. Peer support empowers both the PSW and the service user. While the service user finds empathy and understanding, the PSW finds a renewed sense of purpose and confidence. In this way, peer support would alleviate loneliness and isolation that result from a variety of causes while instilling longer-term resilience into the community.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
A service user refers to any individual who uses the peer support service for assistance.
Peer support work can be empowering and healing for those in recovery. However, as with any emotionally challenging work, PSWs benefit from ongoing support and supervision. Some interactions can be draining, especially if they hit too close to home. It is common for 988 Lifeline call centers to have a trained staff member, such as a psychologist or therapist, on call for such situations. A PSW could use this resource to debrief or process as needed. Additionally, during recruitment, PSW candidates should be screened to ensure they are comfortable speaking about their own lived experience and helping others who are going through the same experience.
Most PSWs would be compensated in the same way as their 988 Lifeline call and text specialist counterparts. They would be compensated with rigorous ongoing training, support, and resources for recovery and self-care in return for their time. They would also benefit from a social support system of fellow PSWs. Some individuals who may be ideal candidates for peer support work may struggle to find employment and health insurance. They will be less likely to volunteer if they do not have a living wage. To maximize the range of lived experiences available, certain individuals should be eligible for financial compensation.
A service user’s phone call or text should be routed to a 988 Lifeline peer support operator or telephone triage, where the service user is asked to say aloud (or type) what kind of support they are searching for (e.g., “I want to speak with someone who has been in prison.”) Then, the service user’s call (or text) would be routed to a PSW (under an alias) anywhere in the United States who 1) is currently on shift and 2) has a lived experience that is closest to the service user’s request. Each PSW would have associated keywords, such as “former incarceration,” “PTSD,” or “living alone,” which means that they are trained to connect with any service user about those lived experiences. The PSW would follow the service user’s lead in the conversation, and the PSW could share parts of their own lived experience when appropriate.
Text messaging can be more accessible than a phone line for youths and people with disabilities. The 988 Lifeline includes a text messaging option for this same reason.
It is true that both the regular 988 Lifeline and peer support service would provide resources and emotional support. By incorporating peer support into the 988 Lifeline, existing local and national peer support organizations would be eligible to partner with the 988 Lifeline and bolster its capacity to provide peer support. This endeavor will help with some staffing concerns. Furthermore, the peer support service would cover a diverse range of lived experiences extending beyond mental health. Therefore, more individuals may be motivated to join the 988 Lifeline staff to share their unique lived experience and help others who feel the same way.
Integrating a versatile peer support service into the 988 Lifeline transforms the latter into a service that every single American can use. (Every American has some unique lived experience.) The service’s versatility may help incentivize the remaining states to pass legislation to collect a 988 Lifeline fee through telephone and wireless service providers.
Finally, peer support is a cost-effective, preventative intervention. It should help remove the burden on other federal services, thereby reducing overall spending in time.
After implementing the action plan outlined in this memo, a subsequent memo should outline how to integrate peer support work into the community in person and on a large scale. Incorporating peer support into the 988 Lifeline would show its effectiveness to the public, policymakers, and healthcare professionals. This credibility would bolster endeavors to integrate peer support work into community settings throughout the country (e.g., behavioral health centers, hospitals and emergency rooms, and community clinics). These endeavors could also lead to professionalizing peer support, so PSWs can be reimbursed through Medicaid programs and health insurance.
Clearing the Path for New Uses for Generic Drugs
The labeling-only 505(b)(2) NDA pathway for non-manufacturers to seek FDA approval
Repurposing generic drugs as new treatments for life-threatening diseases is an exciting yet largely overlooked opportunity due to a lack of market-driven incentives. The low profit margins for generic drugs mean that pharmaceutical companies rarely invest in research, regulatory efforts, and marketing for new uses. Nonprofit organizations and other non-commercial non-manufacturers are increasing efforts to repurpose widely available generic drugs and rapidly expand affordable treatment options for patients. However, these non-manufacturers find it difficult to obtain regulatory approval in the U.S. They face significant challenges in using the existing approval pathways, specifically in: 1) providing the FDA with required chemistry, manufacturing, and controls (CMC) data, 2) providing the FDA with product samples, and 3) conducting post-marketing surveillance. Without a straightforward path for approval and updating drug labeling, non-manufacturers have relied on off-label use of repurposed drugs to drive uptake. This practice results in outdated labeling for generics and hinders widespread clinical adoption, limiting patient access to these potentially life-saving treatments.
To encourage greater adoption of generic drugs in clinical practice – that is, to encourage the repurposing of these drugs – the FDA should implement a dedicated regulatory pathway for non-manufacturers to seek approval of new indications for repurposed generic drugs. A potential solution is an extension of the existing 505(b)(2) new drug application (NDA) approval pathway. This extension, the “labeling-only” 505(b)(2) NDA, would be a dedicated pathway for non-manufacturers to seek FDA approval of new indications for well-established small molecule drugs when multiple generic products are already available. The labeling-only 505(b)(2) pathway would be applicable for repurposing drugs for any disease. Creating a regulatory pathway for non-manufacturers would unlock access to innovative therapies and enable the public to benefit from the enormous potential of low-cost generic drugs.
Challenge and Opportunity
The opportunity for generic drug repurposing
On-patent, branded drugs are often unaffordable for Americans. Due to the high cost of care, 42% of patients in the U.S. exhaust their life savings within two years of a cancer diagnosis. Generic drug repurposing – the process of finding new uses for FDA-approved generic drugs – is a major opportunity to quickly create low-cost and accessible treatment options for many diseases. In oncology, hundreds of generic drugs approved for non-cancer uses have been tested as cancer treatments in published preclinical and clinical studies.
The untapped potential for generic drug repurposing in cancer and other diseases is not being realized because of the lack of market incentives. Pharmaceutical companies are primarily focused on de novo drug development to create new molecular and chemical entities. Typically, pharmaceutical companies will invest in repurposing only when the drugs are protected by patents or statutory market exclusivities, or when modification to the drugs can create new patent protection or exclusivities (e.g., through new formulations, dosage forms, or routes of administration). Once patents and exclusivities expire, the introduction of generic drugs creates competition in the marketplace. Generics can be up to 80-85% less expensive than their branded counterparts, driving down overall drug prices. The steep decline in prices means that pharmaceutical companies have little motivation to invest in research and marketing for new uses of off-patent drugs, and this loss of interest often starts in the years preceding generic entry.
In theory, pharmaceutical companies could repurpose generics without changing the drugs and apply for method-of-use patents, which should provide exclusivity for new indications and the potential for higher pricing. However, due to substitution of generic drugs at the pharmacy level, method-of-use patents are of little to no practical value when there are already therapeutically equivalent products on the market. Pharmacists can dispense a generic version instead of the patent-protected drug product, even if the substituted generic does not have the specific indication in its labeling. Currently, nearly all U.S. states permit substitution by the pharmacy, and over a third have regulations that require generic substitution when available.
Nonprofits like Reboot Rx and other non-commercial non-manufacturers are therefore stepping in to advance the use of repurposed generic drugs across many diseases. Non-manufacturers, which do not manufacture or distribute drugs, aim to ensure there is substantial evidence for new indications of generic drugs and then advocate for their clinical use. Regulatory approval would accelerate adoption. However, even with substantial evidence to support regulatory review, non-manufacturers find it difficult or impossible to seek approval for new indications of generic drugs. There is no straightforward pathway to do so within the current U.S. regulatory framework without offering a specific, manufactured version of the drug. This challenge is not unique to the U.S.; recent efforts in the European Union (EU) have sought to address the regulatory gap. In the 2023 EU reform of pharmaceutical legislation, Article 48 is currently under review by the European Parliament as a potential solution to allow nonprofit entities to spearhead submissions for the approval of new indications for authorized medicinal products with the European Medicines Agency. To maximize the patient impact of generic drugs in America, non-manufacturers should be able to drive updates to FDA drug labeling, enabling widespread clinical adoption of repurposed drugs in a formal, predictable, and systematic manner.
The importance of FDA approval
Drugs that are FDA-approved can be prescribed for any indication not listed on the product labeling, often referred to as “off-label use”. Since non-manufacturers face significant challenges pursuing regulatory approval for new indications, they often must rely on advocating for off-label use of repurposed drugs.
While off-label use is widely accepted and helpful for specific circumstances, there are significant advantages to having FDA approval of new drug indications included in labeling. FDA drug labeling is intended to contain up-to-date information about drug products and ensures that the necessary conditions of use (including dosing, warnings, and precautions) are communicated for the specific indications. It is the primary authoritative source for making informed treatment decisions and is heavily valued by the medical community. Approval may increase the likelihood of uptake by clinical guidelines, pathways systems, and healthcare payers. Indications with FDA approval may generate greater awareness of the treatment options, leading to a broader and more rapid impact on clinical practice.
Clinical practice guidelines are often the leading authority for prescribers and patients regarding off-label use. In oncology, for example, the National Comprehensive Cancer Network (NCCN) Guidelines are commonly used guidelines that include many off-label uses. However, guidelines do not exist for every disease and medical specialty, which can make it more difficult to gain acceptance for off-label uses. The Centers for Medicare and Medicaid Services (CMS) policy routinely covers off-label drug uses if they are listed in certain compendia. The NCCN Compendia, which is based on the NCCN Guidelines, is the only accepted compendium that is disease-specific.
Off-label use requires more effort from individual prescribers and patients to independently evaluate new drug data, thereby slowing uptake of the treatments. This can be especially difficult for community-based physicians, who need to remain up-to-date on new treatment options across many diseases. Off-label prescribing can also introduce medico-legal risks, such as malpractice. These burdens and risks limit off-label prescribing, even when there is supportive evidence for the new uses.
As new uses for generic drugs are discovered, it is crucial to update the labeling to ensure alignment with current clinical practice. Outdated generic drug labeling means that prescribers and patients may not have access to all the necessary information to understand the full risk-benefit profile. Americans deserve to have access to all effective treatment options – especially low-cost and widely available generic drugs that could help mitigate the financial toxicity and health inequities faced by many patients. For the public benefit, the FDA should support approaches that remove regulatory barriers for non-manufacturers and modernize drug labeling.
Existing pathways for manufacturers to obtain FDA approval
The current FDA approval system is based on the idea that sponsors have discrete physical drug products. Traditionally, sponsors seeking FDA approval are pharmaceutical companies or drug manufacturers that intend to produce (or contract for production), distribute, and sell the finished drug product. For the purposes of FDA regulation, “drug” refers to a substance intended for use in the treatment or prevention of disease; “drug product” is the final dosage form that contains a drug substance and inactive ingredients made and sold by a specific manufacturer. One drug can be present on the market in multiple drug products. In the current regulatory framework, drug products are approved through one of the following:
- 505(b)(1) NDA: New drug products, including new molecular entities, are first submitted for FDA review through the 505(b)(1) new drug application (NDA) pathway. In this pathway, the sponsor either generates all necessary clinical data or owns the right of reference to the clinical data needed to support the NDA. FDA approval of an NDA establishes a reference listed drug (RLD), based on the FDA’s determinations of safety and effectiveness for the drug.
- 505(b)(2) NDA: For new drug products or formulations utilizing or related to previously approved drugs or drug substances, the 505(b)(2) NDA pathway offers a streamlined alternative to the 505(b)(1) NDA. By allowing the sponsor to reference the FDA’s findings of safety and effectiveness for previously approved drugs and published literature, the sponsor can submit data they do not own or for which they do not have the right of reference, once any exclusivities expire.
- ANDA: Generic drug equivalents are approved through the 505(j) abbreviated NDA (ANDA) pathway. The RLD serves as the standard that other manufacturers may reference when seeking approval for generic versions of the drug. Upon approval, ANDA products receive a therapeutic equivalence designation in the FDA’s Orange Book, indicating the generic drug products are interchangeable for and substitutable with the RLD.
- BLA: Biological products are approved through the biologics license application (BLA). There is currently no provision analogous to section 505(b)(2) under the law governing the approval of biological products. While the FDA is authorized to approve biosimilars, including interchangeable biosimilars akin to generics, no current pathway permits the addition of new indications for use to a biosimilar product short of submitting a complete, original BLA with full preclinical and clinical data. Our proposed regulatory pathway is relevant for small molecule drugs and would not be applicable to biological products under current law.
Manufacturers can add new indications to their approved labeling without modifying the drug product through existing pathways. With supportive clinical evidence for the new indication, the NDA holder can file a supplemental NDA (sNDA), while an ANDA holder may submit a 505(b)(2) NDA as a supplement to their existing ANDA. As previously discussed, the drug product will likely be subject to pharmacy-level substitution with any available therapeutically equivalent generic. The marketing exclusivities that sponsors may receive from the FDA do not protect against this substitution. Therefore, these pathways are rarely, if ever, used by pharmaceutical companies when there are already multiple generic manufacturers of the product.
Challenges for non-manufacturers in using existing pathways
Since manufacturers are not incentivized to seek regulatory approval for new indications, labeling changes are more likely to happen if driven by non-manufacturers. Yet non-manufacturers face significant challenges in utilizing the existing regulatory pathways. Sponsors must submit the following information for all NDAs for the FDA’s review: 1) clinical and nonclinical data on the safety and effectiveness of the drug for the proposed indication; 2) the proposed labeling; and 3) chemistry, manufacturing, and controls (CMC) data describing the methods of manufacturing and the controls to maintain the drug product’s quality.
To submit and maintain NDAs, non-manufacturer sponsors would need to address the following challenges:
- Providing the FDA with required CMC data. NDA sponsors must provide CMC data for FDA review. Non-manufacturers would not produce physical drug products, and therefore they would not have information on the manufacturing process.
- Providing the FDA with product samples. If requested, NDA sponsors must have the drug products and other samples (e.g., drug substances or reference standards) available to support the FDA review process and must make available for inspection the facilities where the drug substances and drug products are manufactured. Non-manufacturers would not have physical drug products to provide as samples, the capabilities to produce them, or access to the facilities where they are made.
- Conducting post-marketing surveillance. Post-marketing responsibilities to maintain an NDA include conducting annual safety reporting and maintaining a toll-free number for the public to call with questions or concerns. Non-manufacturers, such as small nonprofits, may not have the bandwidth or resources to meet these requirements.
Within the current statutory framework, a non-manufacturer could sponsor a 505(b)(2) NDA to obtain approval of a new indication by partnering with a current manufacturer of the drug – either an NDA or ANDA holder. The manufacturer would help meet the technical requirements of the 505(b)(2) application that the non-manufacturer could not fulfill independently. Through this partnership, the non-manufacturer would acquire the CMC data and physical drug product samples from the manufacturer and rely on the manufacturer’s facilities to fulfill FDA inspection and quality requirements.
Once approved, the 505(b)(2) NDA would create a new drug product with indication-specific labeling, even though the product would be identical to an existing product under a previous NDA or ANDA. The 505(b)(2) NDA would then be tied to the specific manufacturer due to the use of their CMC data, and that manufacturer would be responsible for producing and distributing the drug product for the new indication.
As a practical matter, this pathway is rarely attainable. Manufacturers of marketed drug products, particularly generic drug manufacturers, lack the incentives needed to partner with non-manufacturers. Manufacturers may not want to provide their CMC data or samples because it may prompt FDA inspection of their facilities, require an update to their CMC information, or open the door to product liability risks. The existing incentive structure strongly discourages generic drug manufacturers from expending any additional resources on researching new uses or making any changes to their product labeling that would deviate from the original RLD product.
Plan of Action
To modernize drug labeling and enhance clinical adoption of generic drugs, the FDA should implement a dedicated regulatory pathway for non-manufacturers to seek approval of new indications for repurposed generic drugs. Ultimately, such a pathway would enable drug repurposing and be a crucial step toward equitable healthcare access for Americans. We propose a potential solution – a “labeling-only” 505(b)(2) NDA – as an extension of the existing 505(b)(2) approval pathway.
Overview of the proposed labeling-only 505(b)(2) NDA pathway
The labeling-only 505(b)(2) NDA would enable non-manufacturers to reference CMC information from previous FDA determinations and, when necessary, provide the FDA with samples of commercially available drug products. Through this approach, the new indication would not be tied to a specific drug product made by one manufacturer. There is no inherent necessity for a new indication of a generic drug to be exclusively linked to a single manufacturer or drug product when the FDA has already approved multiple therapeutically equivalent generic drugs. Any of these interchangeable drug products would be considered equally safe and effective for the new indication, and patients could receive any of these drug products due to pharmacy-level substitution.
We describe non-manufacturer repurposing sponsors as entities that intend to submit or reference clinical data through a labeling-only 505(b)(2) NDA. This pathway is designed to expand the FDA-approved labeling of generic drugs for new indications, including those that may already be considered the standard of care. Non-manufacturers do not have the means to independently produce or distribute drug products. Instead, they intend to show that there is substantial evidence to support the new use through FDA approval, and then advocate for the indication in clinical practice. This evidence may be based on their research or research performed by other entities, including clinical trials and real-world data analyses.
The labeling-only 505(b)(2) NDA pathway helps address the three major challenges non-manufacturers face in pursuing regulatory approval. Through this pathway, non-manufacturers would be able to:
- Reference the FDA’s previous determinations on CMC data. Currently, a 505(b)(2) NDA can reference the FDA’s previous determinations of safety and effectiveness for an approved drug product. For eligible generic drugs, the labeling-only 505(b)(2) NDA would build on this practice by allowing non-manufacturer sponsors to reference the FDA’s previous determinations on any NDA or ANDA that the manufacturing process and CMC data are adequate to meet regulatory standards.
- Provide the FDA with product samples using commercially available drug product samples. Currently, it is up to the discretion of the FDA whether or not to request samples in the review of an application. With the labeling-only 505(b)(2) NDA, non-manufacturers would provide the FDA with samples of commercially available products from generic manufacturers. Given that the FDA would have already evaluated the products and their bioequivalence to the RLD during the previous reviews, it is not expected that the FDA would need to re-examine the product at the level of requesting samples, except potentially to examine the packaging and physical presentation of the product for compatibility with the new indication and conditions of use. The facilities where the drugs are made would remain available for inspection, under the same terms and conditions as the existing, approved marketing applications.
- Manage post-marketing responsibilities. Since most post-marketing surveillance and adverse event reporting are drug product-specific, these obligations would continue to be the responsibility of the manufacturer of the physical drug product dispensed. With the labeling-only 505(b)(2) NDA, the non-manufacturer would not have product-specific obligations because they are not putting a new product into the marketplace. However, we anticipate the non-manufacturer would be responsible for the repurposed indication on their labeling, including but not limited to post-marketing surveillance as well as indication-specific adverse event reporting and reasonable follow-up.
Under the labeling-only 505(b)(2) NDA, the non-manufacturer sponsor would not introduce a new physical drug product into the market. The new labeling created by the approval would not expressly be associated with one specific product. The non-manufacturer’s labeling would refer to the drug by its established generic name. In that way, the non-manufacturer sponsor’s approval and labeling could be applicable to all equivalent versions of the drug product and would be available for patients to receive from their pharmacy in the same way that generic drugs are typically dispensed at the pharmacy. That is, with the benefit of pharmacy-level substitution, patients could receive any available, therapeutically equivalent drug products from any current manufacturer.
Eligibility criteria
We envision the users of this pathway to be non-manufacturers that conduct drug repurposing research for the public benefit, including organizations like nonprofits and patient advocacy groups. The FDA should implement and enforce additional guardrails on eligibility to ensure that sponsors operate in good faith and cannot otherwise meet traditional NDA requirements. This process may include pre-submission meetings and reviews. The labeling-only 505(b)(2) NDA should be held to the FDA’s standard level of rigor and scrutiny of safety and effectiveness for the proposed indication during the review process.
The labeling-only 505(b)(2) would only be suitable for well-established, commercially available small molecule generic drugs, which can be identified as:
- Drugs with a U.S. Pharmacopeia and National Formulary (USP-NF) monograph. The USP-NF monograph system ensures the uniformity of available products on the market by setting a consensus minimum standard of identity, strength, quality, and purity among all marketed versions of a drug. It is expressly recognized in the Federal Food, Drug, and Cosmetics Act (FDCA). The USP-NF strives to have substance and product monographs for all FDA-approved drugs. USP-NF monographs for generics are commonly available because the drugs have been on the market for a long time and are typically produced by multiple manufacturers. Drug products in the U.S. market must conform to the standards in the USP-NF, when available, to avoid possible charges of adulteration and misbranding.
By statute and regulation, the FDA already allows for NDAs and ANDAs to reference the USP-NF to satisfy some CMC requirements, such as for specifications of the drug substance. As an illustration of the acceptance of the USP-NF, clinical trial protocols requiring the use of background therapy or supportive care, as well as trials testing medical devices requiring the use of a drug product, often will specify that any available version of the drug product meeting USP-NF standards can be used. We propose that products without USP-NF monographs, including certain newer drugs and drugs with especially complicated manufacturing processes that are not conducive to standardization, would not be eligible for the labeling-only 505(b)(2) pathway.
- Drugs with multiple A-rated, therapeutically equivalent products in the FDA Orange Book. The FDA does not regulate which specific products are dispensed or substituted for a given drug prescription. The listing of therapeutic equivalents in the Orange Book facilitates the seamless replacement of drug products from different manufacturers in clinical practice. Therapeutically equivalent drug products: i) have demonstrated bioequivalence to the RLD; ii) have the same strength, dosage form, and route of administration as the RLD; and iii) are labeled for the same conditions of use as the RLD. Therapeutic equivalents that meet these criteria are designated “A-rated” in the Orange Book. A-rated drug products are substitutable for any other version of that A-rated drug product, including the RLD itself.
Implementation
The labeling-only 505(b)(2) NDA pathway could be implemented through an FDA guidance document interpreting the current statute and regulations or through legislation that clarifies the FDA’s existing authority. Guidance documents contain the FDA’s interpretation of a given policy on regulatory issues. The FDA’s Center for Drug Evaluation and Research (CDER) could issue new guidance that allows for interpretation of the existing statute, thereby officially authorizing previous FDA determinations of acceptable CMC data to be referenceable for eligible generic drugs and adjusting drug sample requirements. Alternatively, the labeling-only 505(b)(2) could be enacted by Congress through a statutory change by incorporation into FDCA, which is up for reauthorization through the Prescription Drug User Fee Act (PDUFA) in 2027, or other congressional acts as appropriate. FDA guidance would be a faster pathway to adoption, while statutory authorization would offer additional safeguards for the continuance of the pathway long term.
The labeling-only 505(b)(2) NDA pathway could be funded through user fees, which are established by PDUFA for the cost to file and maintain NDAs. However, many nonprofit sponsors would not be able to afford the same user fees as for-profit pharmaceutical manufacturers. Relevant statutes will likely need to be updated to create a different fee schema for non-manufacturers who use the labeling-only 505(b)(2) pathway. In a similar spirit to reducing barriers to maintaining up-to-date labeling, the 2017 PDUFA update waived the fee for submitting an sNDA, which is how an existing RLD holder would update their labeling with new indication information.
Conclusion
Patients need new and affordable treatment options for diseases that have a devastating societal impact, and repurposing generic drugs can help address this need. Nonprofits and other non-manufacturers are driving these efforts forward due to a lack of interest from pharmaceutical companies. As momentum gains for generic drug repurposing, the U.S. regulatory system needs a pathway for non-manufacturers to seek FDA approval of new indications for existing generic drugs. Our proposed labeling-only 505(b)(2) NDA would eliminate undue administrative burden, enabling non-manufacturers to pursue FDA approval of new indications. It would allow the FDA to provide the public with the most up-to-date drug labeling, improving the ability of patients and physicians to make informed treatment decisions. This dedicated pathway would increase the availability of effective treatment options while reducing costs for the American healthcare system.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
The FDA does not have sufficient bandwidth or resources to meet the opportunity we have with repurposed generic drugs. To be the primary driver of labeling changes for repurposed generic drugs, the FDA would need to identify repurposing opportunities and also thoroughly compile and evaluate the safety and effectiveness data for the new indications. Project Renewal in FDA’s Oncology Center of Excellence is working with RLD holders to update the labeling of certain older oncology drugs where the outdated labeling does not reflect their current clinical use. The initial focus of Project Renewal is limited, and newly repurposed treatments are not included within its scope. For newly repurposed treatments, the FDA could evaluate the drugs and post to the Federal Register reports on their safety and effectiveness that could be referenced by manufacturers. However, this approach is burdensome as it would require a significant commitment of FDA resources. By introducing a motivated, third-party non-manufacturer as the primary driver for labeling changes, non-manufacturers can contribute expertise and resources to enable faster data evaluation for more drugs.
The pre-existing NDA sponsor could update their labeling to add the new indication through an sNDA that references the labeling-only 505(b)(2) NDA. ANDA holders would then be legally required to match their labeling to that of the RLD. The FDA should determine whether all current manufacturers would be required to update their labeling following approval of the new indication, and if so, the appropriate process.
Generic drugs play a vital role in the U.S. healthcare system by decreasing drug spending and increasing the accessibility of essential medicines. Generics account for 91% of prescriptions filled in the U.S. Expanding the market with generics for new indications could lead to short-term price increases above the inflation rate for off-patent branded and generic drugs in some unlikely circumstances. For example, if a new use for a generic drug substantially increases demand for the drug, there is a short-term risk that prices for the drug or potential substitutes could rise until manufacturers build more capacity to increase supply. To mitigate this risk, generic manufacturers could be notified about potential increases in demand so they can plan for increased production.
Generally, healthcare payers are not required to cover or reimburse for off-label uses of drugs. Unless a drug undergoes utilization management, payers cover most generic drugs for off-label uses because coverage is agnostic of indication. Clinical practice guidelines are highly influential in the widespread adoption of off-label treatments into the standard of care and are often referenced by payers making reimbursement decisions. In oncology, many off-label treatments are included in guidelines; only 62% of treatments in the NCCN are aligned with FDA-approved indications. For example, more than half of the NCCN recommendations for metastatic breast cancer are off-label treatments. Due to the breadth of off-label use, we anticipate payers would continue to cover repurposed generic drugs used off-label, even if there is a pathway available for non-manufacturers to pursue FDA approval.
We do not envision any form of exclusivity being granted for indications pursued via a labeling-only 505(b)(2) NDA. Given that the non-manufacturer sponsor would rely on existing products produced by multiple generic manufacturers, there is no new product to grant exclusivity. Even if some form of exclusivity were given to the non-manufacturer, it would be insufficient to guarantee the use of any particular drug product over another due to pharmacy-level substitution.
An Innovation Agenda for Addiction: Breakthrough Medicines That Scale
The federal government should expand the FDA’s priority review voucher program (PRV) and provide market exclusivity advantages to encourage the development of medications for addiction.
Taken together, substance use disorders (alcohol, cigarettes, and other drugs) cause more deaths in the U.S. every year than cancer or heart disease and cause devastating downstream social harms. Despite this, only 3% of eligible patients received substance use disorder (SUD) medication, a result of low uptake and efficacy of existing medications and a lack of options for patients addicted to stimulants. This is due to a near-total absence of pharmaceutical research and development activity. To make population level impact to reduce harms from opioids, methamphetamine, cocaine, alcohol, and cigarettes, we must address the broken market dynamics in addiction medicine.
The PRV program should be expanded to cover opioid use disorder, alcohol use disorder, stimulant use disorder, and smoking. In addition, drugs that are approved for these SUD indications should have extended exclusivity and sponsors that develop these medications should receive vouchers to extend exclusivity for other medications.
Challenge and Opportunity
Addiction policy efforts on both the left and the right have struggled. Despite substantial progress reducing smoking, 29 million Americans still smoke cigarettes and feel unable to quit and 480,000 Americans die each year from smoking. While overdose deaths from opioids, cocaine, and methamphetamine have fallen slightly from their peak in 2022, they are still near record highs, three times higher than 20 years ago. Alcohol deaths per capita have doubled since 1999.
Roughly 60% of all crimes and 65% of violent crimes are related to drugs or alcohol; and the opioid crisis alone costs the United States $1.5 trillion a year. Progress in reducing addiction is held back because people with a substance use disorder take medication. This low uptake has multiple causes: in opiate use disorder, uptake is persistently low despite recent relaxations of prescription rules, with patients reporting a variety of reasons for refusal; treatments for alcohol use disorder have modest effects; and there are no approved treatments for stimulant use disorder. Only three percent take SUD medications, as shown in figure 1 below [link to image]. In brief, only 2% of those suffering alcohol use disorder, 13% of those with opiate use disorder, 2% of smokers, and approximately 0% of illicit stimulant users are receiving medication, giving a weighted average of about 3%.
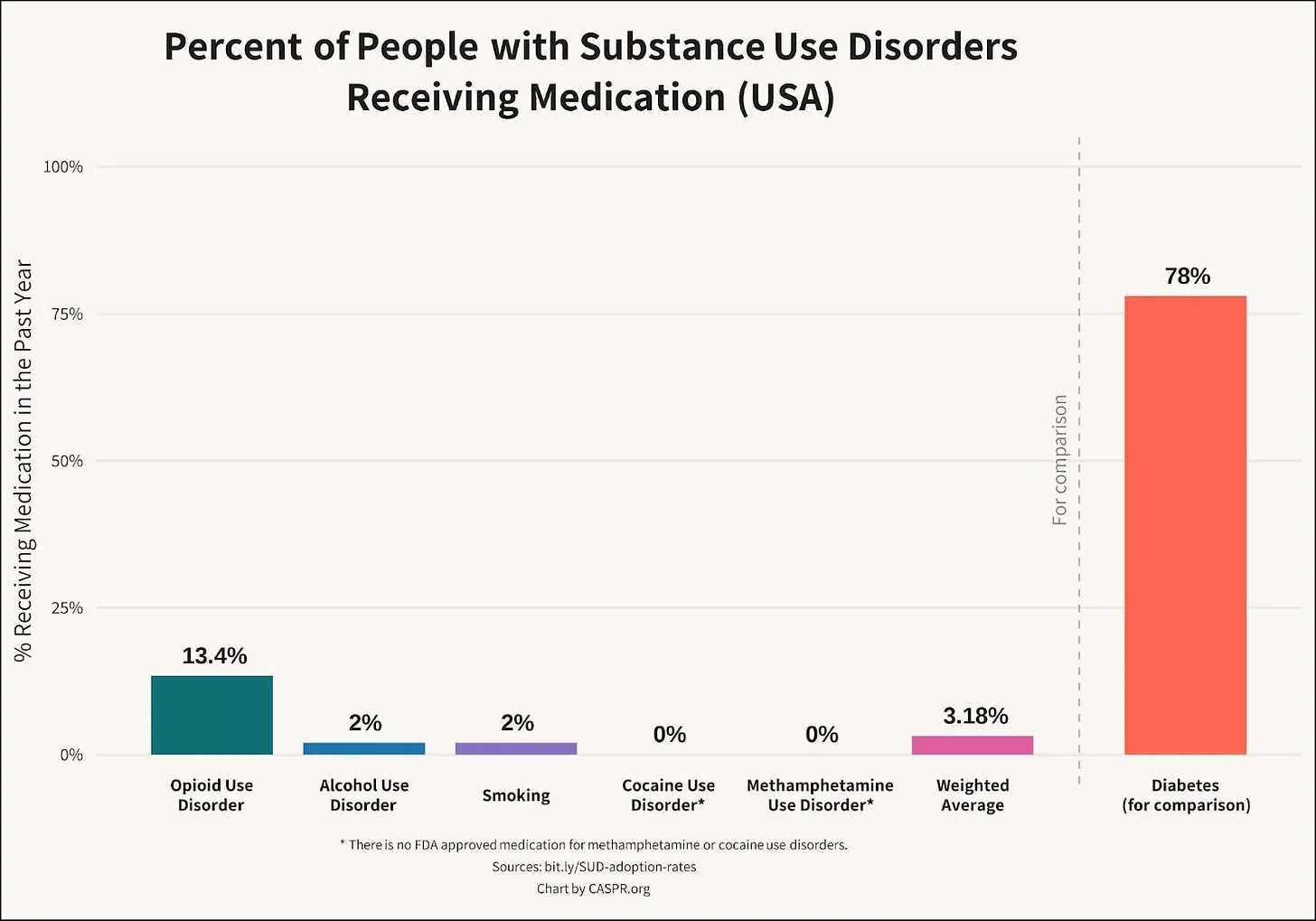{kind=link}
There has been rapid innovation in the illicit market as synthetic opioids and expanded meth production have lowered price and increased strength and availability. Meanwhile, there has been virtually no innovation in medicines to prevent and treat addiction. The last significant FDA approval for opioid use disorder was buprenorphine in 2002; progress since then has been minimal, with new formulations or dosing of old medications. For alcohol use disorder, the most recent was acamprosate in 2004 (and it is rarely prescribed due to limited efficacy and three times a day dosing).
None of the ten largest pharmaceutical companies have active addiction medicine programs or drug candidates, and the pharmaceutical industry as a whole has only pursued minimal drug development. According to the trade association BIO, “Venture investment into companies with novel addiction drug programs over the last 10 years is estimated at $130M, 270 times less than oncology.”
There are promising addiction drug candidates being studied by academics but without industry support they will never become medicines. If pharmaceutical companies spent just 10% of what they spend on obesity therapies, we might quickly make progress.
For example, GLP-1 medicines like Ozempic and Mounjaro have strong anti-addictive effects across substances. Randomized trials and real-world patient health record studies show dramatic drops in consumption of drugs and alcohol for patients taking a GLP-1. Many addiction scientists now consider these compounds to be the biggest breakthrough in decades. However, Novo Nordisk and Eli Lilly, who own the drugs currently in the market, do not plan to run phase 3 addiction trials on them, due to fear of adverse events in substance use disorder populations. The result is that a huge medical opportunity is stuck in limbo indefinitely. Fortunately, Lilly has recently signaled that they will run trials on related compounds, but remain years from approval.
Conversations with industry leaders make clear that large pharmas avoid SUD indications for several reasons. First, their upside appears limited, since current SUD medications have modest sales. Second, like other psychiatric disorders, the problem is challenging given the range and complexity of neurological targets and the logistical challenges of recruiting people with substance use disorder as participants. Finally, companies face downside reputational and regulatory risk if participants, who face high baseline rates of death from overdose regardless, were to die in trials. In the case of Ozempic and Mounjaro, sponsors face an obstacle some have termed the “problem of new uses” – clinical trials of an already lucrative drug for a new indication carry downside risk if new side effects or adverse events are reported.
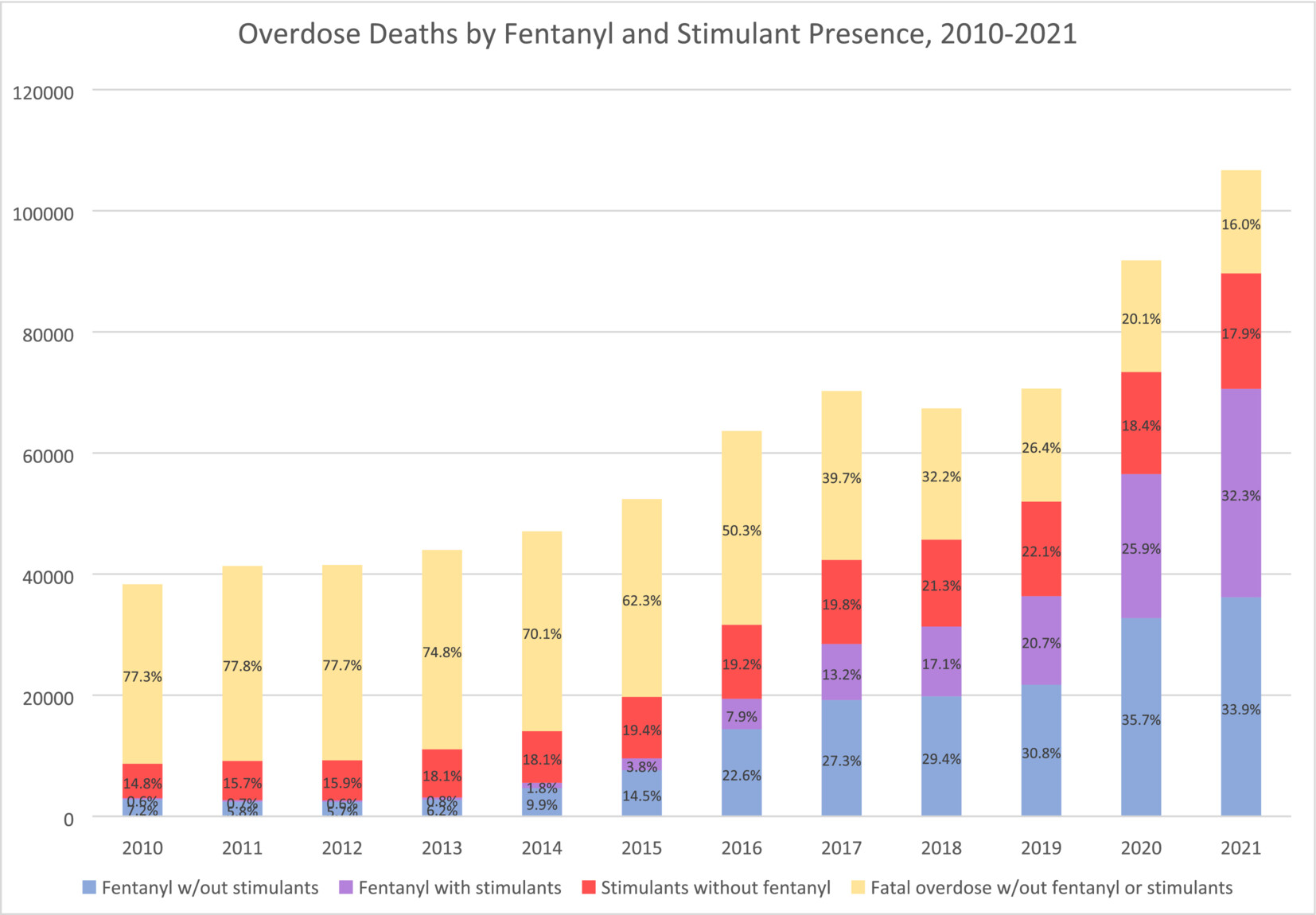
Image from Charting the fourth wave, based on CDC data
Plan of Action
Market Shaping Interventions
Recommendation 1. Expand the FDA priority review voucher (PRV) program to include addiction medicine indications.
The FDA priority review voucher (PRV) program incentivizes development of drugs for rare pediatric and infectious diseases by rewarding companies who get drugs approved with a transferable voucher that accelerates FDA approval. These vouchers are currently selling for an average of $100M. The PRV program doesn’t cost the government any money but it makes drug development in the designated categories much more lucrative. The PRV program has proven very successful, leading to a surge in approvals of medications.
As a neglected market with urgent unmet medical and public health needs, and which also promises to benefit the broader public by reducing the negative externalities of addiction, addiction medicine is a perfect fit for the PRV program. Doing so could transform the broken market dynamics of the field. The advantage of the PRV program is that it does not require substantial new congressional appropriations, though it will require Congress giving the FDA authority to expand the PRV program, as it has done previously to add other disease areas.
Recommendation 2. Extend exclusivity for addiction medicines and incentivize pursuit of new indications
Market exclusivity is a primary driver of pharmaceutical industry revenue. Extending exclusivities would have a very large effect on industry behavior and is needed to create sufficient incentives. The duration of exclusivity for alcohol use disorder, opioid use disorder, stimulant use disorder, and smoking cessation indications should be extended along with other incentives.
- Addiction medicine indications should receive an additional two years of exclusivity for biologics and three years for small molecules.
- Companies that achieve an indication for a substance use disorder for a medication that represents a significant advance would receive an exclusivity voucher that can be transferred to another medication. For 2nd, 3rd, and 4th SUD indications with the same compound, companies would be granted a shorter duration exclusivity voucher. Durations would be tiered, as described in this proposal from Duke, to balance public interest and reward levels.
- FDA should provide increased consideration for addiction medicines for breakthrough, fast track, and priority review designations, as well as accelerate meeting schedules, all of which substantially reduce development expenses.
For precedent, there are already a number of FDA programs that extend medication exclusivity, including ‘orphan drug exclusivity’ and the qualified infectious disease product (QIDP) program. Like rare diseases and antibiotics, addiction is a market that requires incentives to function effectively. In addition, successful treatments, given the negative externalities of addiction, have public benefit beyond the direct medical impact, and deserve additional public incentives.
Recommendation 3. Modernize FDA Standards of Efficacy for Substance Use Disorder Trials
A significant barrier to pharmaceutical innovation in SUDs is outdated or unpredictable efficacy standards sometimes set by the FDA for clinical trials. Efficacy expectations for substance use disorder indications are often rooted in abstinence-only and other binary measure orientations that the scientific and medical community has moved past when evaluating substance use disorder harms.
This article in the American Journal of Drug and Alcohol Abuse demonstrates that binary outcome measures like ‘number of heavy drinking days’ (NHDD) can underestimate the efficacy of treatments. This recent report from NIAAA on alcohol trial endpoints recommends a shift away from abstinence-based endpoints and towards more meaningful consumption-based endpoints. This approach should be adopted by the FDA for all SUD treatments, not just alcohol.
There are some indications that the FDA has begun modernizing their approach. This recent paper from NIH and FDA on smoking cessation therapies provides updated guidance that moves in the right direction.
More broadly, the FDA should work to adopt endpoints and standards of efficacy that mirror standards in other disease areas. This shift is best achieved through new guidance or statements issued by the FDA, which would offer positive assurance to pharmaceutical companies that they have achievable paths to approval. Predictability throughout the medication development life cycle is absolutely essential for companies considering investment.
Congress should include statements in upcoming appropriations and authorizations that state:
- The FDA should adopt non-binary standards of efficacy for addiction treatments that are aligned with standards for other common disorders and the FDA shall, within 12 months, report on the standards employed for substance use disorder relative to other prevalent chronic conditions and report steps to eliminate disparities in evidentiary standards and issue new guidance on the subject.
- The FDA should publish clear guidance on endpoints across SUDs to support planning among pharmaceutical companies considering work in this field.
Conclusion
Sustained focus and investment in diabetes and heart disease treatments has enabled medical breakthroughs. Addiction medicine, by contrast, has been largely stagnant for decades. Stimulating private-sector interest in addiction medicine through regulatory and exclusivity incentives, as well as modernized efficacy standards, is essential for disrupting the status quo. Breakthroughs in addiction medicine could save hundreds of thousands of lives in the US and provide long-term relief for one of our most intractable social problems. Given the negative externalities of addiction, this would also have enormous benefits for society at large, reducing crime and intergenerational trauma and saving money on social services and law enforcement.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
Per author conversations with industry leaders, private sector interest in SUD medication development is limited for the following reasons:
- The upside of pursuing SUD indications appears limited, since current SUD medications, which are generally targeted for specific substances, have modest sales.
- Even with preliminary evidence that GLP-1 drugs may be efficacious for some SUD indications (e.g, alcohol, opiates, and tobacco), companies are reluctant to pursue label expansion for SUD. As described previously, with already lucrative drugs, companies face a downside risk (termed the “problem of new uses”) from running large clinical trials, and possibly uncovering new side effects or incurring random adverse events which could harm reputation and existing markets.
- In the specific case of SUD, this downside risk might be especially large, since people with substance use disorder have high baseline rates of overdose and death.
Moreover, there is an argument that a treatment for SUD is a public good, to the degree that it ameliorates the negative externalities of addiction – increasing the case for more public-sector incentives for SUD treatment. The end result is that medical treatments for SUD are stuck in an indefinite limbo, with private-sector interest in SUD, as documented previously, being very low.
The current lack of effective and widely used SUD medications is disheartening, but this is in the context of private sector disinterest and scant funding. Even modest successes in SUD treatment have the potential to kickstart an innovation loop, akin to the rush of biotech companies hastening to enter the obesity treatment field. Prior to the success of the GLP-1 drugs, obesity treatment had been moribund, and viewed pessimistically in light of drugs that had limited efficacy or had been withdrawn for side effects like suicidality or cardiovascular issues.
An SUD success like GLP-1 for obesity has the potential to kindle a similar rush of interest; the challenge is the initiation of that cascade. Given the very low levels of investment in SUD treatments, there is potential low-hanging fruit that, given sufficient funding, could be trialed and deployed.
There has been rapid innovation in the field of addiction, but it’s been happening on the wrong side: addiction-inducing technologies are becoming more powerful, while SUD treatments have largely stagnated. This innovation is most evident in synthetic opioids and methamphetamine.
Compared to heroin, fentanyl is about 25x stronger (on a per-weight basis), and hence, much easier to smuggle. As the Commission on Combating Synthetic Opioid Trafficking put it:
Single-digit metric tonnage of pure fentanyl is not a large amount and could easily fit into a shipping container or a truck trailer, which seriously challenges interdiction…Perhaps as much as 5 MT [metric tons] of pure fentanyl would be needed to satisfy the entire annual U.S. consumption for illegally supplied opioids.
Moreover, as a recent Scientific American article documented, innovations in fentanyl production, including the use of safer precursors and methods that don’t require sophisticated equipment, mean that fentanyl production is now decentralized, and resistant to attempts by law enforcement to shut it down.
As fentanyl has come to dominate the opioid supply over the past 10 years, overdose deaths have risen dramatically. New synthetic opioids and non-opioids like xylazine are also becoming common.
At the same time, due to advances in production techniques in Mexico, methamphetamine production has skyrocketed in recent decades while purity has improved. Worst of all, unlike heroin, fentanyl is easily combined with meth and cocaine in pills and powder.
The DEA has highlighted the presence of “super labs” in Mexico capable of producing hundreds of pounds of meth per batch.
Together, these three innovations (fentanyl, cheap meth, and new combinations) have led to a 400% increase in overdose deaths in the past 20 years. Without equally powerful innovations to reduce addiction rates, we will never make long-term and sustainable progress.
From Strategy to Impact: Establishing an AI Corps to Accelerate HHS Transformation
To unlock the full potential of artificial intelligence (AI) within the Department of Health and Human Services (HHS), an AI Corps should be established, embedding specialized AI experts within each of the department’s 10 agencies. HHS is uniquely positioned for—and urgently requires—this investment in AI expertise, as it plays a pivotal role in delivering efficient healthcare to millions of Americans. HHS’s responsibilities intersect with areas where AI has already shown great promise, including managing vast healthcare datasets, accelerating drug development, and combating healthcare fraud.
Modeled after the success of the Department of Homeland Security (DHS)’s existing AI Corps, this program would recruit top-tier professionals with advanced expertise in AI, machine learning, data science, and data engineering to drive innovation within HHS. While current HHS initiatives like the AI Council and AI Community of Practice provide valuable strategic guidance, they fall short in delivering the on-the-ground expertise necessary for meaningful AI adoption across HHS agencies. The AI Corps would fill this gap, providing the hands-on, agency-level support necessary to move beyond strategy and into the impactful implementation intended by recent federal actions related to AI.
This memo uses the Food and Drug Administration (FDA) as a case study to demonstrate how an AI Corps member could spearhead advancements within HHS’s agencies. However, the potential benefits extend across the department. For instance, at the Centers for Disease Control and Prevention (CDC), AI Corps experts could leverage machine learning for more precise outbreak modeling, enabling faster, more targeted public health responses. At the National Institutes of Health (NIH), they could accelerate biomedical research through AI-driven analysis of large-scale genomic and proteomic data. Similarly, at the Centers for Medicare and Medicaid Services (CMS), they could improve healthcare delivery by employing advanced algorithms for patient data analytics, predicting patient outcomes, and enhancing fraud detection mechanisms.
Challenge and Opportunity
AI is poised to revolutionize not only healthcare but also the broad spectrum of services under HHS, offering unprecedented opportunities to enhance patient outcomes, streamline administrative processes, improve public health surveillance, and advance biomedical research. Realizing these benefits and defending against potential harms demands the effective implementation and support of AI tools across HHS. The federal workforce, though committed and capable, currently lacks the specialized expertise needed to fully harness AI’s potential, risking a lag in AI adoption that could impede progress.
The public sector is responding well to this opportunity since it is well positioned to attract leading experts to help leverage new technologies. However, for federal agencies, attracting technical experts has been a perennial challenge, resulting in major setbacks in government tech projects: Of government software projects that cost more than $6 million, only 13% succeed.
Without introducing a dedicated AI Corps, existing employees—many of whom lack specialized AI expertise—would be required to implement and manage complex AI tools alongside their regular duties. This could lead to the acquisition or development of AI solutions without proper evaluation of their suitability or effectiveness for specific use cases. Additionally, without the necessary expertise to oversee and monitor these systems, agencies may struggle to ensure they are functioning correctly and ethically. As a result, there could be significant inefficiencies, missed opportunities for impactful AI applications, and an increased reliance on external consultants who may not fully understand the unique challenges and needs of each agency. This scenario not only risks undermining the effectiveness of AI initiatives but also heightens the potential for errors, biases, and misuse of AI technologies, ultimately hindering HHS’s mission and objectives.
HHS’s AI Strategy recognizes the need for AI expertise in government; however, its focus has largely been on strategic oversight rather than the operational execution needed on the ground, with the planned establishment of an AI Council and AI Community of Practice prioritizing policy and coordination. While these entities are crucial, they do not address the immediate need for hands-on expertise within individual agencies. This leaves a critical gap in the hands-on expertise required to safely implement AI solutions at the agency level. HHS covers a wide breadth of functions, from administering national health insurance programs like Medicare and Medicaid to conducting advanced biomedical research at the NIH, with each agency facing distinct challenges where AI could provide transformative benefits. However, without dedicated support, AI adoption risks becoming fragmented, underutilized, or ineffective.
For example, at the CDC, AI could significantly improve infectious disease surveillance systems, enabling more timely interventions and enhancing the CDC’s overall preparedness for public health crises, moving beyond traditional methods that often rely on slower, manual analysis. Furthermore, the Administration for Children and Families (ACF) could leverage AI to better allocate resources, improve program outcomes, and support vulnerable populations more effectively. There are great opportunities to use machine learning algorithms to accelerate data processing and discovery in fields such as cancer genomics and personalized medicine. This could help researchers identify new biomarkers, optimize clinical trial designs, and push forward breakthroughs in medical research faster and more efficiently. However, without the right expertise, these game-changing opportunities could not only remain unrealized but also introduce significant risks. The potential for biased algorithms, privacy breaches, and misinterpretation of AI outputs poses serious concerns. Agency leaders may feel pressured to adopt technologies they don’t fully understand, leading to ineffective or even harmful implementations. Embedding AI experts within HHS agencies is essential to ensure that AI solutions are deployed responsibly, maximizing benefits while mitigating potential harms.
This gap presents an opportunity for the federal government to take decisive action. By recruiting and embedding top-tier AI professionals within each agency, HHS could ensure that AI is treated not as an ancillary task but as a core component of agency operations. These experts would bring the specialized knowledge necessary to integrate AI tools safely and effectively, optimize processes, and drive innovation within each agency.
DHS’s AI Corps, launched as part of the National AI Talent Surge, provides a strong precedent for recruiting AI specialists to advance departmental capabilities. For instance, AI Corps members have played a vital role in improving disaster response by using AI to quickly assess damage and allocate resources more effectively during crises. They have also enhanced cybersecurity efforts by using AI to detect vulnerabilities in critical U.S. government systems and networks. Building on these successes, a similar effort within HHS would ensure that AI adoption moves beyond a strategic objective to a practical implementation, with dedicated experts driving innovation across the department’s diverse functions.
Case Study: The Food and Drug Administration (FDA)
The FDA stands at the forefront of the biotechnology revolution, facing the dual challenges of rapid innovation and a massive influx of complex data. Advances in gene editing, personalized medicine, and AI-driven diagnostics promise to transform healthcare, but they also present significant regulatory hurdles. The current framework, though robust, struggles to keep pace with these innovations, risking delays in the approval and implementation of groundbreaking treatments.
This situation is reminiscent of the challenges faced in the 1980s and 1990s, when advances in pharmaceutical science outstripped the FDA’s capacity to review new drugs, leading to the so-called “drug lag.” The Prescription Drug User Fee Act of 1992 was a pivotal response, streamlining the drug review process by providing the FDA with additional resources. However, the continued reliance on scaling resources may not be sustainable as the complexity and volume of data increase.
The FDA has begun to address this new challenge. For example, the Center for Biologics Evaluation and Research has established committees like the Artificial Intelligence Coordinating Committee and the Regulatory Review AI Subcommittee. However, these efforts largely involve existing staff who must balance AI responsibilities with their regular duties, limiting the potential impact. Moreover, the focus has predominantly been on regulating AI rather than leveraging it to enhance regulatory processes.
Placing an AI expert from the HHS AI Corps within the FDA could fundamentally change this dynamic. By providing dedicated, expert support, the FDA could accelerate its regulatory review processes, ensuring timely and safe access to innovative treatments. The financial implications are significant: the value of accelerated drug approvals, as demonstrated by the worth of Priority Review Vouchers (acceleration of four months = ~$100 million), indicates that effective AI adoption could unlock billions of dollars in industry value while simultaneously improving public health outcomes.
Plan of Action
To address the challenges and seize the opportunities outlined earlier, the Office of the Chief Artificial Intelligence Officer (OCAIO) within HHS should establish an AI Corps composed of specialized experts in artificial intelligence, machine learning, data science, and data engineering. This initiative will be modeled after DHS’s successful AI Corps and tailored to the unique needs of HHS and its 10 agencies.
Recommendation 1. Establish an AI Corps within HHS.
Composition: The AI Corps would initially consist of 10 experts hired to temporary civil servant positions, with one member allocated to each of HHS’s 10 agencies, and each placement lasting one to two years. These experts will possess a range of technical skills—including AI, data science, data engineering, and cloud computing—tailored to each agency’s specific needs and technological maturity. This approach ensures that each agency has the appropriate expertise to effectively implement AI tools and methodologies, whether that involves building foundational data infrastructure or developing advanced AI applications.
Hiring authority: The DHS AI Corps utilized direct hiring authority, which was expanded by the Office of Personnel Management under the National AI Talent Surge. HHS’s AI Corps could adopt a similar approach. This authority would enable streamlined recruitment of individuals into specific AI roles, including positions in AI research, machine learning, and data science. This expedited process would allow HHS to quickly hire and onboard top-tier AI talent.
Oversight: The AI Corps would be overseen by the OCAIO, which would provide strategic direction and ensure alignment with HHS’s broader AI initiatives. The OCAIO would also be responsible for coordinating the activities of the AI Corps, setting performance goals, and evaluating outcomes.
Budget and Funding
Estimated cost: The AI Corps is projected to cost approximately $1.5 million per year, based on an average salary of $150,000 per corps member. This estimate includes salaries and operational costs such as training, travel for interagency collaboration, and participation in conferences.
Funding source: Funding would be sourced from the existing HHS budget, specifically from allocations set aside for digital transformation and innovation. Given the relatively modest budget required, reallocation within these existing funds should be sufficient.
Recruitment and Training
Selection process: AI Corps members would be recruited through a competitive process, targeting individuals with proven expertise in AI, data science, and related fields.
Training: Upon selection, AI Corps members would undergo an intensive orientation and training program to familiarize them with the specific needs and challenges of HHS’s various agencies. This also includes training on federal regulations, ethics, and data governance to ensure that AI applications comply with existing laws and policies.
Agency Integration
Deployment: Each AI Corps member would be embedded within a specific HHS agency, where they would work closely with agency leadership and staff to identify opportunities for AI implementation. Their primary responsibility would be to develop and deploy AI tools that enhance the agency’s mission-critical processes. For example, an AI Corps member embedded at the CDC could focus on improving disease surveillance systems through AI-driven predictive analytics, while a member at the NIH could drive advancements in biomedical research by using machine learning algorithms to analyze complex genomic data.
Collaboration: To ensure cross-agency learning and collaboration, AI Corps members would convene regularly to share insights, challenges, and successes. These convenings would be aligned with the existing AI Community of Practice meetings, fostering a broader exchange of knowledge and best practices across the department.
Case Study: The FDA
AI Corps Integration at the FDA
Location: The AI Corps member assigned to the FDA would be based in the Office of Digital Transformation, reporting directly to the chief information officer. This strategic placement would enable the expert to work closely with the FDA’s leadership team, ensuring that AI initiatives are aligned with the agency’s overall digital strategy.
Key responsibilities
Process improvement: The AI Corps member would collaborate with FDA reviewers to identify opportunities for AI to streamline regulatory review processes. This might include developing AI tools to assist with data analysis, automate routine tasks, or enhance decision-making capabilities.
Opportunity scoping: The expert would engage with FDA staff to understand their workflows, challenges, and data needs. Based on these insights, the AI Corps member would scope and propose AI solutions tailored to the FDA’s specific requirements.
Pilot projects: The AI Corps member would lead pilot projects to test AI tools in real-world scenarios, gathering data and feedback to refine and scale successful initiatives across the agency.
Conclusion
Establishing an AI Corps within HHS is a critical step toward harnessing AI’s full potential to enhance outcomes and operational efficiency across federal health agencies. By embedding dedicated AI experts within each agency, HHS can accelerate the adoption of innovative AI solutions, address current implementation gaps, and proactively respond to the evolving demands of the health landscape.
While HHS may currently have less technological infrastructure compared to departments like the Department of Homeland Security, targeted investment in in-house expertise is key to bridging that gap. The proposed AI Corps not only empowers agencies like the FDA, CDC, NIH, and CMS to enhance their missions but also sets a precedent for effective AI integration across the federal government. Prompt action to establish the AI Corps will position HHS at the forefront of technological innovation, delivering tangible benefits to the American public and transforming the way it delivers services and fulfills its mission.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
The AI Corps is designed to be the opposite of bureaucracy—it’s about action, not administration. These experts will be embedded directly within agencies, working alongside existing teams to solve real-world problems, not adding paperwork. Their mission is to integrate AI into daily operations, making processes more efficient and outcomes more impactful. By focusing on tangible results and measurable improvements, the AI Corps will be judged by its ability to cut through red tape, not create it.
Innovation can present challenges, but the AI Corps is designed to address them effectively. These experts will not only bring technical expertise but also serve as facilitators who can translate advanced AI capabilities into practical applications that align with existing agency cultures. A key part of their role will be to make AI more accessible and understandable, ensuring it is valuable to all levels of staff, from frontline workers to senior leadership. Their success will depend on their ability to seamlessly integrate advanced technology into the agency’s everyday operations.
AI isn’t just another tool; it’s a force multiplier that can help solve those other pressing issues more effectively. Whether it’s accelerating drug approvals at the FDA or enhancing public health responses across HHS, AI has the potential to improve outcomes, save time, and reduce costs. By embedding AI experts within agencies, we’re not just addressing one problem—we’re empowering the entire department to tackle multiple challenges with greater efficiency and impact.
For top AI talent, the AI Corps offers a unique opportunity to make a difference at a scale that few private-sector roles can match. It’s a chance to apply their skills to public service, tackling some of the nation’s most critical challenges in healthcare, regulation, and beyond. The AI Corps members will have the opportunity to shape the future of AI in government, leaving a legacy of innovation and impact. The allure of making a tangible difference in people’s lives can be a powerful motivator for the right kind of talent.
While outsourcing AI talent or using consultants can offer short-term benefits, it often lacks the sustained engagement necessary for long-term success. Building in-house expertise through the AI Corps ensures that AI capabilities are deeply integrated into the agency’s operations and culture. A notable example illustrating the risks of overreliance on external contractors is the initial rollout of HealthCare.gov. The website faced significant technical issues at launch due to coordination challenges and insufficient in-house technical oversight, which hindered public access to essential healthcare services. In contrast, recent successful government initiatives—such as the efficient distribution of COVID-19 test kits and the timely processing of economic stimulus payments directly into bank accounts—demonstrate the positive impact of having the right technical experts within government agencies.
Collaboration is crucial to the AI Corps’ success. Instead of working in isolation, AI Corps members will integrate with existing IT and data teams, bringing specialized AI knowledge that complements the teams’ expertise. This partnership approach ensures that AI initiatives are well-grounded in the agencies’ existing infrastructure and aligned with ongoing IT projects. The AI Corps will serve as a catalyst, amplifying the capabilities of existing teams rather than duplicating their efforts.
The AI Corps is focused on augmentation, not replacement. The primary goal is to empower existing staff with advanced tools and processes, enhancing their work rather than replacing them. AI Corps members will collaborate closely with agency employees to automate routine tasks and free up time for more meaningful activities. A 2021 study by the Boston Consulting Group found that 60% of employees view AI as a coworker rather than a replacement. This reflects the intent of the AI Corps—to build capacity within agencies and ensure that AI is a tool that amplifies human effort, fostering a more efficient and effective workforce.
Success for the AI Corps program means that each HHS agency has made measurable progress toward integrating AI and related technologies, tailored to their specific needs and maturity levels. Within one to two years, agencies might have established robust data infrastructures, migrated platforms to the cloud, or developed pilot AI projects that address key challenges. Success also includes fostering a culture of innovation and experimentation, with AI Corps members identifying opportunities and creating proofs of concept in low-risk environments. By collaborating across agencies, these experts support each other and amplify the program’s impact. Ultimately, success is reflected in enhanced capabilities and efficiencies within agencies, setting a strong foundation for ongoing technological advancement aligned with each agency’s mission.
Creating an HHS Loan Program Office to Fill Critical Gaps in Life Science and Health Financing
We propose the establishment of a Department of Health and Human Services Loan Programs Office (HHS LPO) to fill critical and systematic gaps in financing that prevent innovative life-saving medicines and other critical health technologies from reaching patients, improving health outcomes, and bolstering our public health. To be effective, the HHS LPO requires an authority to issue or guarantee loans, up to $5 billion in total. Federally financed debt can help fill critical funding gaps and complement ongoing federal grants, contracts, reimbursement, and regulatory policies and catalyze private-sector investment in innovation.
Challenge and Opportunity
Despite recent advances in the biological understanding of human diseases and a rapidly advancing technological toolbox, commercialization of innovative life-saving medicines and critical health technologies face enormous headwinds. This is due in part to the difficulty in accessing sustained financing across the entire development lifecycle. Further, macroeconomic trends such as non-zero interest rates have substantially reduced deployed capital from venture capital and private equity, especially with longer investment horizons.
The average new medicine requires 15 years and over $2 billion to go from the earliest stages of discovery to widespread clinical deployment. Over the last 20 years, the earliest and riskiest portions of the drug discovery process have shifted from the province of large pharmaceutical companies to a patchwork of researchers, entrepreneurs, venture capitalists, and supporting organizations. While this trend has enabled new entrants into the biotechnology landscape, it has also required startup companies to navigate labyrinthine processes of technical regulatory guidelines, obtaining long-term and risk-friendly financing, and predicting payor and provider willingness to ultimately adopt the product.
Additionally, there are major gaps in healthcare infrastructure such as lack of adequate drug manufacturing capacity, healthcare worker shortages, and declining rural hospitals. Limited investment is available for critical infrastructure to support telehealth, rural healthcare settings, biomanufacturing, and decentralized clinical trials, among others.
The challenges in health share some similarities to other highly regulated, capital-intensive industries, such as energy. The Department of Energy (DOE) Loan Program Office (LPO) was created in 2005 to offer loans and loan guarantees to support businesses in deploying innovative clean energy, advanced transportation, and tribal energy projects in the United States. LPO has closed more than $40 billion in deals to date. While agencies across HHS rely primarily on grants and contracts to deploy research and development (R&D) funding, capital-intensive projects are best deployed as loans, not only to appropriately balance risk between the government and lendees but also to provide better stewardship over taxpayer resources via mechanisms that create liquidity with lower budget impact. Moreover, private-sector financing rates are subject to market-based interest rates, which can have enormous impacts on available capital for R&D.
Plan of Action
There are many federal credit programs across multiple departments and agencies that provide a strong blueprint for the HHS LPO to follow. Examples include the aforementioned DOE Loan Programs Office, which provides capital to scale large-scale energy infrastructure projects using new technologies, and the Small Business Administration’s credit programs, which provide credit financing to small businesses via several loan and loan matching programs.
Proposed Actions
We propose the following three actions:
- Pass legislation authorizing the HHS LPO to make or guarantee loans supporting the development of critical medicines that serve unmet or underserved public health needs.
- Establish the LPO as a staffing division within HHS.
- Appropriate a budget to support the staffing and operations of the newly created HHS LPO.
Scope
Similar to how DOE LPO services the priorities of the DOE, the HHS LPO would develop strategy priorities based on market gaps and public health gaps. It would also develop a rigorous diligence process to prioritize, solicit, assess, and manage potential deals, in alignment with the Federal Credit Reform Act and the associated policies set forth by the Office of Management and Budget and followed by all federal credit programs. It would also seek companion equity investors and creditors from the private sector to create leverage and would provide portfolio support via demand-alignment and -generation mechanisms (e.g., advance manufacturing commitments and advanced market commitments from insurers).
We envision several possible areas of focus for the HHS LPO:
- Providing loans or loan guarantees to amplify investment funds that use venture capital or other private investment tools, such as early-stage drug development or biomanufacturing capacity. While these funds may already exist, they are typically underpowered.
- Providing large-scale financing in partnership with private investors to fund major healthcare infrastructure gaps, such as rural hospitals, decentralized clinical trial capacity, telehealth services, and advanced biomanufacturing capacity.
- Providing financing to test new innovative finance models, e.g. portfolio-based R&D bonds, designed to attract additional capital into under-funded R&D and lower financial risks.
Conclusion
To address the challenges in bringing innovative life-saving medicines and critical health technologies to market, we need an HHS Loan Programs Office that would not only create liquidity by providing or guaranteeing critical financing for capital-intensive projects but address critical gaps in the innovation pipeline, including treatments for rare diseases, underserved communities, biomanufacturing, and healthcare infrastructure. Finally, it would be uniquely positioned to pilot innovative financing mechanisms in partnership with the private sector to better align private capital towards public health goals.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
The DOE LPO, enabled via the Energy Policy Act of 2005, enables the Secretary of Energy to provide loan guarantees toward publicly or privately financed projects involving new and innovative energy technologies.
The DOE LPO provides a bridge to private financing and bankability for large-scale, high-impact clean energy and supply chain projects involving new and innovative technologies. It also expands manufacturing capacity and energy access within the United States. The DOE LPO has enabled companies involving energy and energy manufacturing technologies to achieve infrastructure-scale growth, including Tesla, an electric car manufacturer; Lithium Americas Corp., a company supplying lithium for batteries; and the Agua Caliente Solar Project, a solar power station sponsored by NRG Solar that was the largest in the world at its time of construction.
The HHS LPO would similarly augment, guarantee, or bridge to private financing for projects involving the development and deployment of new and innovative technologies in life sciences and healthcare. It would draw upon the structure and authority of the DOE LPO as its basis.
The HHS LPO could look to the DOE LPO for examples as to how to structure potential funds or use cases. The DOE LPO’s Title 17 Clean Energy Financing Program provides four eligible project categories: (1) projects deploying new or significantly improved technology; (2) projects manufacturing products representing new or significantly improved technologies; (3) projects receiving credit or financing from counterpart state-level institutions; and (4) projects involving existing infrastructure that also share benefits to customers or associated communities.
Drawing on these examples, the HHS LPO could support project categories such as (1) emerging health and life science technologies; (2) the commercialization and scaling access of novel technologies; and (3) expanding biomanufacturing capacity in the United States, particularly for novel platforms (e.g., cell and gene therapies).
The budget could be estimated via its authority to make or guarantee loans. Presently, the DOE LPO has over $400 billion in loan authority and is actively managing a portfolio of just over $30 billion. Given this benchmark and the size of the private market for early-stage healthcare venture capital valued at approximately $20 billion, we encourage the creation of an HHS LPO with $5 billion in loan-making authority. Using proportional volume to the $180 million sought by DOE LPO in FY2023, we estimate that an HHS LPO with $5 billion in loan-making authority would require a budget appropriation of $30 million.
The HHS LPO would be subject to oversight by the HHS Inspector General, OMB, as well as the respective legislative bodies, the House of Representatives Energy and Commerce Committee and the Senate Health, Education, Labor and Pension Committee.
Like the DOE LPO, the HHS LPO would publish an Annual Portfolio Status Report detailing its new investments, existing portfolio, and other key financial and operational metrics.
It is also possible for Congress to authorize existing funding agencies, such as BARDA, the Advanced Research Projects Agency for Health (ARPA-H), or the National Institutes for Health (NIH), with loan authority. However, due the highly specialized talent needed to effectively operate a complex loan financing operation, the program is significantly more likely to succeed if housed into a dedicated HHS LPO that would then work closely with the other health-focused funding agencies within HHS.
The other alternative is to expand the authority for other LPOs and financing agencies, such as the DOE LPO or the U.S. Development Finance Corporation, to focus on domestic health. However, that is likely to create conflicts of priority given their already large and diverse portfolios.
The project requires legislation similar to the Department of Energy’s Title 17 Clean Energy Financing Program, created via the Energy Policy Act of 2005 and subsequently expanded via the Infrastructure Investment and Jobs Act in 2021 and the Inflation Reduction Act in 2022.
This legislation would direct the HHS to establish an office, presumably a Loan Programs Office, to make loan guarantees to support new and innovative technologies in life sciences and healthcare. While the LPO could reside within an existing HHS division, the LPO would most ideally be established in a manner that enables it to serve projects across the full Department, including those from the National Institutes of Health, Food and Drug Administration, Biomedical Advanced Research and Development Authority, and the Centers for Medicare and Medicaid Services. As such, it would preferably not reside within any single one of these organizations. Like the DOE LPO, the HHS LPO would be led by a director, who would be directed to hire the necessary finance, technical, and operational experts for the function of the office.
Drawing on the Energy Policy Act of 2005 that created the DOE LPO, enabling legislation for the HHS Loan Programs office would direct the Secretary of HHS to make loan guarantees in consultation with the Secretary of Treasury toward projects involving new and innovative technologies in healthcare and life sciences. The enabling legislation would include several provisions:
- Necessary included terms and conditions for loan guarantees created via the HHS LPO, including loan length, interest rates, and default provisions;
- Allowance of fees to be captured via the HHS LPO to provide funding support for the program; and
- A list of eligible project types for loan guarantees.
Supporters are likely to include companies developing and deploying life sciences and healthcare technologies, including early-stage biotechnology research companies, biomanufacturing companies, and healthcare technology companies. Similarly, patient advocates would be similarly supportive because of the LPO’s potential to bring new technologies to market and reduce the overall Weighted Average Cost of Capital (WACC) for biotechnology companies, potentially supporting expanded access.
Existing financiers of research in biomedical sciences technology may be neutral or ambivalent toward this policy. On one hand, it would provide expanded access to syndicated financing or loan guarantees that would compound the impact of each dollar invested. On the other hand, most financiers currently use equity financing, which enables the demand for a high rate of return via subsequent investment and operation. An LPO could provide financing that requires a lower rate of return, thereby diluting the impact of current financiers in the market.
Skeptics are likely to include groups opposing expansions of government spending, particularly involving higher-risk mechanisms like loan guarantees. The DOE LPO has drawn the attention of several such skeptics, oftentimes leading to increased levels of oversight from legislative stakeholders. The HHS LPO could expect similar opposition. Other skeptics may include companies with existing medicines and healthcare technologies, who may be worried about competitors introducing products with prices and access provisions that have been enabled via financing with lower WACC.