Terminal Patients Need Better Access to Drugs and Clinical Trial Information
Editor’s note: This policy memo was written by Jake Seliger and his wife Bess Stillman. Jake passed away before meeting his child, Athena, born on October 31, 2024. Except where indicated, the first-person voice is that of Jake. This memo advocates for user-centric technology modernization and data interoperability. More crucially, he urges expanded patient rights to opt-in to experimental drug trials and FDA rule revisions to enable terminal patients to take more risks on behalf of themselves, for the benefit of others.
The FDA is supposed to ensure that treatments are safe and effective, but terminal cancer patients like me are already effectively dead. If I don’t receive advanced treatment quickly, I will die. My hope, in the time I have remaining, is to promote policies that will speed up access to treatments and clinical trials for cancer patients throughout the U.S.
There are about two million cancer diagnoses and 600,000 deaths annually in the United States. Cancer treatments are improved over time via the clinical trial system: thousands of clinical trials are conducted each year (many funded by the government via the National Institutes of Health, or NIH, and many funded by pharmaceutical companies hoping to get FDA approval for their products).
But the clinical trial system is needlessly slow, and as discussed below, is nearly impossible for any layperson to access without skilled consulting. As a result, clinical trials are far less useful than they could be.
The FDA is currently “protecting” me from being harmed or killed by novel, promising, advanced cancer treatments that could save or extend my life, so that I can die from cancer instead. Like most patients, I would prefer a much faster system in which the FDA conditionally approves promising, early-phase advanced cancer treatments, even if those treatments haven’t yet been proven fully effective. Drugmakers will be better incentivized to invest in novel cancer treatments if they can begin receiving payment for those treatments sooner. The true risks to terminal patients like me are low—I’m already dying—and the benefits to both existing terminal patients and future patients of all kinds are substantial.
I would also prefer a clinical trial system that was easy for patients to navigate, rather than next-to-impossible. Easier access for patients could radically lower the cost and time for clinical trials by making recruitment far cheaper and more widespread, rather than including only about 6% of patients. In turn, speeding up the clinical-trial process means that future cancer patients will be helped by more quickly approving novel treatments. About half of pharmaceutical R&D spending goes not to basic research, but to the clinical trial process. If we can cut the costs of clinical trials by streamlining the process to improve access to terminal patients, more treatments will make it to patients, and will be faster in doing so.
Cancer treatment is a non-partisan issue. To my knowledge, both left and right agree that prematurely dying from cancer is bad. Excess safety-ism and excessive caution from the FDA costs lives, including in the near future, my own. Three concrete actions would improve clinical research, particular for terminal cancer patients like me, but for many other patients as well:
Clinical trials should be easier and cheaper. The chief obstacles to this are recruitment and retention.
Congress and NIH should modernize the website ClinicalTrials.gov and vastly expand what drug companies and research sites are required to report there, and the time in which they must report it. Requiring timely updates that include comprehensive eligibility criteria, availability for new participants, and accurate site contact information,would mean that patients and doctors will have much more complete information about what trials are available and for whom.
The process of determining patient eligibility and enrolling in a trial should be easier. Due to narrow eligibility criteria, studies struggle to enroll an adequate number of local patients, which severely delays trial progression. A patient who wishes to participate in a trial must “establish care” with the hospital system hosting the trial, before they are even initially screened for eligibility or told if a slot is available. Due to telemedicine practice restrictions across state lines, this means that patients who aren’t already cared for at that site— patients who are ill and for whom unnecessary travel is a huge burden— must use their limited energy to travel to a site just to find out if they can proceed to requesting a trial slot and starting further eligibility testing. Then, if approved for the study, they must be able to uproot their lives to move to, or spend extensive periods of time at, the study location
Improved access to remote care for clinical trials would solve both these problems. First, by allowing the practice of telemedicine across state lines for visits directly related to screening and enrollment into clinical trials. Second, by incentivizing decentralization—meaning a participant in the study can receive the experimental drug and most monitoring, labs and imaging at a local hospital or infusion clinic—by accepting data from sites that can follow a standardized study protocol.
We should require the FDA to allow companies with prospective treatments for fatal diseases to bring those treatments to market after initial safety studies, with minimal delays and with a lessened burden for demonstrating benefit.
Background
[At the time of writing this] I’m a 40-year-old man whose wife is five months pregnant, and the treatment that may keep me alive long enough to meet my child is being kept from me because of current FDA policies. That drug may be in a clinical trial I cannot access. Or, it may be blocked from coming to market by requirements for additional testing to further prove efficacy that has already been demonstrated.
Instead of giving me a chance to take calculated risks on a new therapy that might allow me to live and to be with my family, current FDA regulations are choosing for me: deciding that my certain death from cancer is somehow less harmful to me than taking a calculated, informed risk that might save or prolong my life. Who is asking the patients being protected what they would rather be protected from? The FDA errs too much on the side of extreme caution around drug safety and efficacy, and that choice leads to preventable deaths.
One group of scholars attempted to model how many lives are lost versus gained from a more or less conservative FDA. They find that “from the patient’s perspective, the approval criteria in [FDA program accelerators] may still seem far too conservative.” Their analysis is consistent with the FDA being too stringent and slow in approving use of drugs for fatal diseases like cancer: “Our findings suggest that conventional standards of statistical significance for approving drugs may be overly conservative for the most deadly diseases.”
Drugmakers also find it difficult to navigate what exactly the FDA wants: “their deliberations are largely opaque—even to industry insiders—and the exact role and weight of patient preferences are unclear.” This exacerbates the difficulty drugmakers face in seeking to get treatments to patients faster. Inaction in the form of delaying patient access to drugs is killing people. Inaction is choosing death for cancer patients.
I’m an example of this; I was diagnosed with squamous cell carcinoma (SCC) of the tongue in Oct. 2022. I had no risk factors, like smoking or alcoholism, that put me at risk for SCC. The original tumor was removed in October 2022, and I then had radiation that was supposed to cure me. It didn’t, and the cancer reappeared in April 2023. At that point, I would’ve been a great candidate for a drug like MCLA-158, which has been stuck in clinical trials, despite “breakthrough therapy designation” by the FDA and impressive data, for more than five years. This, despite the fact that MCLA-158 is much easier to tolerate than chemotherapy and arrests cancer in about 70% of patients. Current standard of care chemotherapy and immunotherapy has a positive response rate of only 20-30%.
Had MCLA-158 been approved, I might have received it in April 2023, and still have my tongue. Instead, in May 2023, my entire tongue was surgically removed in an attempt to save my life, forever altering my ability to speak and eat and live a normal life. That surgery removed the cancer, but two months later it recurred again in July 2023. While clinical-trial drugs are keeping me alive right now, I’m dying in part because promising treatments like MCLA-158 are stuck in clinical trials, and I couldn’t get them in a timely fashion, despite early data showing their efficacy. Merus, the maker of MCLA-158, is planning a phase 3 trial for MCLA-158, despite its initial successes. This is crazy: head and neck cancer patients need MCLA-158 now.

I’m only writing this brief because I was one of the few patients who was indeed, at long last, able to access MCLA-158 in a clinical trial, which incredibly halted my rapidly expanding, aggressive tumors. Without it, I’d have been dead nine months ago. Without it, many other patients already are. Imagine that you, or your spouse, or parent, or child, finds him or herself in a situation like mine. Do you want the FDA to keep testing a drug that’s already been shown to be effective and allow patients who could benefit to suffer and die? Or do you want your loved one to get the drug, and avoid surgical mutilation and death? I know what I’d choose.
Multiply this situation across hundreds of thousands of people per year and you’ll understand the frustration of the dying cancer patients like me.
As noted above, about 600,000 people die annually from cancer—and yet cancer drugs routinely take a decade or more to move from lab to approval. The process is slow: “By the time a drug gets into phase III, the work required to bring it to that point may have consumed half a decade, or longer, and tens if not hundreds of millions of dollars.” If you have a fatal disease today, a treatment that is five to ten years away won’t help. Too few people participate in clinical trials partly because participation is so difficult; one study finds that “across the entire U.S. system, the estimated participation rate to cancer treatment trials was 6.3%.” Given how many people die, the prospect of life is attractive.
There is another option in between waiting decades for a promising drug to come to market and opening the market to untested drugs: Allowing terminal patients to consent to the risk of novel, earlier-phase treatments, instead of defaulting to near-certain death, would potentially benefit those patients as well as generate larger volumes of important data regarding drug safety and efficacy, thus improving the speed of drug approval for future patients. Requiring basic safety data is reasonable, but requiring complete phase 2 and 3 data for terminal cancer patients is unreasonably slow, and results in deaths that could be prevented through faster approval.
Again, imagine you, your spouse, parent, or child, is in a situation like mine: they’ve exhausted current standard-of-care cancer treatments and are consequently facing certain death. New treatments that could extend or even save their life may exist, or be on the verge of existing, but are held up by the FDA’s requirements that treatments be redundantly proven to be safe and highly effective. Do you want your family member to risk unproven but potentially effective treatments, or do you want your family member to die?
I’d make the same choice. The FDA stands in the way.
Equally important, we need to shorten the clinical trial process: As Alex Telford notes, “Clinical trials are expensive because they are complex, bureaucratic, and reliant on highly skilled labour. Trials now cost as much as $100,000 per patient to run, and sometimes up to $300,000 or even $500,000 per patient.” And as noted above, about half of pharmaceutical R&D spending goes not to basic research, but to the clinical trial process.
Cut the costs of clinical trials, and more treatments will make it to patients, faster. And while it’s not reasonable to treat humans like animal models, a lot of us who have fatal diagnoses have very little to lose and consequently want to try drugs that may help us, and people in the future with similar diseases. Most importantly, we understand the risks of potentially dying from a drug that might help us and generate important data, versus waiting to certainly die from cancer in a way that will not benefit anybody. We are capable of, and willing to give, informed consent. We can do better and move faster than we are now. In the grand scheme of things, “When it comes to clinical trials, we should aim to make them both cheaper and faster. There is as of yet no substitute for human subjects, especially for the complex diseases that are the biggest killers of our time. The best model of a human is (still) a human.” Inaction will lead to the continued deaths of hundreds of thousands of people annually.
Trials need patients, but the process of searching for a trial in which to enroll is archaic. We found ClinicalTrials.gov nearly impossible to navigate. Despite the stakes, from the patient’s perspective, the clinical trial process is impressively broken, obtuse and confusing, and one that we gather no one likes: patients don’t, their families don’t, hospitals and oncologists who run the clinical trials don’t, drug companies must not, and the people who die while waiting to get into a trial probably don’t.
I [Bess] knew that a clinical trial was Jake’s only chance. But how would we find one? I’d initially hoped that a head and neck oncologist would recommend a specific trial, preferably one that they could refer us to. But most doctors didn’t know of trial options outside their institution, or, frequently, within it, unless they were directly involved. ,Most recommended large research institutions that had a good reputation for hard cases, assuming they’d have more studies and one might be a match.
How were they, or we, supposed to find out what trials actually existed?
The only large-scale search option is ClinicalTrials.gov. But many oncologists I spoke with don’t engage with ClinicalTrials.gov, because the information is out-of-date, difficult to navigate, and inaccurate. It can’t be relied on. For example, I shared a summary of Jake’s relevant medical information (with his permission) in a group of physicians who had offered to help us with the clinical trial search. Ten physicians shared their top-five search results. Ominously, none listed the same trials.
How is it that ten doctors can put in the same basic, relevant clinical data into an engine meant to list and search for all existing clinical trials, only for no two to surface the same study? The problem is simple: There’s a lack of keyword standardization.
Instead of a drop-down menu or click-boxes with diagnoses to choose from, the first search “filter” on ClinicalTrials.gov is a text box that says “Condition\Disease.” If I search: “Head and Neck Cancer” I get ___________ results. If I search “Tongue Cancer,” I get _________ results. Although Tongue Cancer is a subset of Head and Neck Cancer, I don’t see the studies listed as “Head and Neck Cancer”, unless I type in both, or the person who created the ClinicalTrials.gov post for the study chose to type out multiple variations on a diagnosis. Nothing says they have to. If I search for both, I will still miss studies filed as: “HNSCC” or “All Solid Tumors” or “Squamous Cell Carcinoma of the Tongue.”
The good news is that online retailers solved this problem for us years ago. It’s easier to find a dress to my exact specifications out of thousands on H&M,com than it is to find a clinical trial. I can open a search bar, click “dress,” select my material from another click box (which allows me to select from the same options the people listing the garments chose from), then click on the boxes for my desired color, dry clean or machine wash, fabric, finish, closure, and any other number of pre-selected categories before adding additional search keywords if I choose. I find a handful of options all relevant to my desires within a matter of minutes. H&M provides a list of standardized keywords describing what they are offering, and I can filter from there. This way, H&M and I are speaking the same language. And a dress isn’t life or death. For much more on my difficulties with ClinicalTrials.gov, see here.
Further slowing a patient’s ability to find a relevant clinical trial is a lack of comprehensive, searchable, eligibility criteria. Every study has eligibility criteria, and eligibility criteria—like keywords—aren’t standardized on ClinicalTrials.gov. Nor is it required that an exhaustive explanation of eligibility criteria be provided, which may lead to a patient wasting precious weeks attempting to establish care and enroll in a trial, only to discover there was unpublished eligibility criteria they don’t meet. Instead, the information page for each study outlines inclusion and exclusion criteria using whatever language whoever is typing feels like using. Many have overlapping inclusion and exclusion criteria, but there can be long lists of additional criteria for each arm of a study, and it’s up to the patient or their doctor to read through them line by line—if they’re even listed— to see if prior medications, current medications, certain genomic sequencing findings, numbers of lines of therapy, etc. makes the trial relevant.
In the end, we hired a consultant (Eileen), who leveraged her full-time work helping pharmaceutical companies determine which novel compounds might be worth pouring their R&D efforts into assisting patients find potential clinical trials. She helped us narrow it down to the top 5 candidate trials from the thousands that turned up in initial queries.
- NCT04815720: Pepinemab in Combination With Pembrolizumab in Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-B84)
- NCT03526835: A Study of Bispecific Antibody MCLA-158 in Patients With Advanced Solid Tumors.
- NCT05743270: Study of RP3 in Combination With Nivolumab and Other Therapy in Patients With Locoregionally Advanced or Recurrent SCCHN.”
- NCT03485209: Efficacy and Safety Study of Tisotumab Vedotin for Patients With Solid Tumors (innovaTV 207)
- NCT05094336: AMG 193, Methylthioadenosine (MTA) Cooperative Protein Arginine Methyltransferase 5 (PRMT5) Inhibitor, Alone and in Combination With Docetaxel in Advanced Methylthioadenosine Phosphorylase (MTAP)-Null Solid Tumors (MTAP).
Based on the names alone, you can see why it would be difficult if not impossible for someone without some expertise in cancer oncology to evaluate trials. Even with Eileen’s expertise, two of the trials were stricken from our list when we discovered unlisted eligibility criteria, which excluded Jake. When exceptionally motivated patients, the oncologists who care for them, and even consultants selling highly specialized assistance can’t reliably navigate a system that claims to be desperate to enroll patients into trials, there is a fundamental problem with the system. But this is a mismatch we can solve, to everyone’s benefit.
Plan of Action
We propose three major actions. Congress should:
Recommendation 1. Direct the National Library of Medicine (NLM) at the National Institutes of Health (NIH) to modernize ClinicalTrials.gov so that patients and doctors have complete and relevant information about available trials, as well as requiring more regular updates from companies as to all the details of available trials, and
Recommendation 2. Allow the practice of telemedicine across state lines for visits related to clinical trials.
Recommendation 3. Require the FDA to allow companies with prospective treatments for fatal diseases to bring those treatments to market after initial safety studies.
Modernizing ClinicalTrials.gov will empower patients, oncologists, and others to better understand what trials are available, where they are available, and their up-to-date eligibility criteria, using standardized search categories to make them more easily discoverable. Allowing telemedicine across state lines for clinical trial care will significantly improve enrollment and retention. Bringing treatments to market after initial safety studies will speed the clinical trial process, and get more patients treatments, sooner. In cancer, delays cause death.
To get more specific:
The FDA already has a number of accelerated approval options. Instead of the usual “right to try” legislation, we propose creating a second, provisional market for terminal patients that allows partial approval of a drug while it’s still undergoing trials, making it available to trial-ineligible (or those unable to easily access a trial) patients for whom standard of care doesn’t provide a meaningful chance at remission. This partial approval would ideally allow physicians to prescribe the drug to this subset of patients as they see fit: be that monotherapy or a variety of personalized combination therapies, tailored to a patient’s needs, side-effect profile and goals. This wouldn’t just expand access to patients who are otherwise out of luck, but can give important data about real-world reaction and response.
As an incentive, the FDA could require pharmaceutical companies to provide drug access to terminal patients as a condition of continuing forward in the trial process on the road to a New Drug Application. While this would be federally forced compassion, it would differ from “compassionate use.” To currently access a study drug via compassionate use, a physician has to petition both the drug company and the FDA on the patient’s behalf, which comes with onerous rules and requirements. Most drug companies with compassionate use programs won’t offer a drug until there’s already a large amount of compelling phase 2 data demonstrating efficacy, a patient must have “failed” standard of care and other available treatments, and the drug must (usually) be given as a monotherapy. Even if the drugmaker says yes, the FDA can still say no. Compassionate use is an option available to very small numbers, in limited instances, and with bureaucratic barriers to overcome. Not terribly compassionate, in my opinion.
The benefits of this “terminal patient” market can and should go both ways, much as lovers should benefit from each other instead of trying to create win-lose situations. Providing the drug to patients would come with reciprocal benefits to the pharmaceutical companies. Any physician prescribing the drug to patients should be required to report data regarding how the drug was used, in what combination, and to what effect. This would create a large pool of observational, real-world data gathered from the patients who aren’t ideal candidates for trials, but better represent the large subset of patients who’ve exhausted multiple lines of therapies yet aren’t ready for the end. Promising combinations and unexpected effects might be identified from these observational data sets, and possibly used to design future trials.
Dying patients would get drugs faster, and live longer, healthier lives, if we can get better access to both information about clinical trials, and treatments for diseases. The current system errs too much on proving effectiveness and too little on the importance of speed itself, and of the number of people who die while waiting for new treatments. Patients like me, who have fatal diagnoses and have already failed “standard of care” therapies, routinely die while waiting for new or improved treatments. Moreover, patients like me have little to lose: because cancer is going to kill us anyway, many of us would prefer to roll the dice on an unproven treatment, or a treatment that has early, incomplete data showing its potential to help, than wait to be killed by cancer. Right now, however, the FDA does not consider how many patients will die while waiting for new treatments. Instead, the FDA requires that drugmakers prove both the safety and efficacy of treatments prior to allowing any approval whatsoever.
As for improving ClinicalTrials.gov, we have several ideas:
First, the NIH should hire programmers with UX experience, and should be empowered to pay market rates to software developers with experience at designing websites for, say, Amazon or Shein. Clinicaltrials.gov was designed to be a study registry, but needs an overhaul to be more useful to actual patients and doctors.
Second, UX is far from the only problem. Data itself can be a problem. For example, the NIH could require a patient-friendly summary of each trial; it could standardize the names of drugs and conditions so that patients and doctors could see consistent and complete search results; and it could use a consistent and machine-readable format for inclusion and exclusion criteria.
We are aware of one patient group that, in trying to build an interface to ClinicalTrials.gov, found a remarkable degree of inconsistency and chaos: “We have analyzed the inclusion and exclusion criteria for all the cancer-related trials from clinicaltrials.gov that are recruiting for interventional studies (approximately 8,500 trials). Among these trials, there are over 1,000 ways to indicate the patient must not be pregnant during a trial, and another 1,000 ways to indicate that the patient must use adequate birth control.” ClinicalTrials.gov should use a Domain Specific Language that standardizes all of these terms and conditions, so that patients and doctors can find relevant trials with 100x less effort.
Third, the long-run goal should be a real-time, searchable database that matches patients to studies using EMR data and allows doctors to see what spots are available and where. Pharmaceutical and biotech companies would need to be required to contribute up-to-date information on all extant clinical trials on a regular basis (e.g., monthly).
Conclusion
Indeed, we need a national clinical trial database that EMRs can connect to, in the style of Epic’s Care Everywhere. A patient signs a release to share their information, like Care Everywhere allows hospital A to download a patient’s information from hospital B if the patient signs a release. Trials with open slots could mark themselves as “recruiting” and update in real time. A doctor could press a button and identify potential open studies. A patient available for a trial could flag their EMR profile as actively searching, allowing clinical trial sites to browse patients in their region who are looking for a study. Access to that patient’s EMR would allow them to scan for eligibility.
This would be easy to do. OKCupid can do it. The tech exists. Finding a clinical trial already feels a bit like online dating, except if you get the wrong match, you die.
This memo produced as part of the Federation of American Scientists and Good Science Project sprint. Find more ideas at Good Science Project x FAS
The FDA has created a variety of programs that have promising-sounding names: “Currently, four programs—the fast track, breakthrough therapy, accelerated approval, and priority review designations—provide faster reviews and/or use surrogate endpoints to judge efficacy. However, published descriptions […] do not indicate any differences in the statistical thresholds used in these programs versus the standard approval process, nor do they mention adapting these thresholds to the severity of the disease.” The problem is that these programs do not appear to do much to actually accelerate getting drugs to patients. MCLA-158 is an example of the problem: the drug has been shown to be safe and effective, and yet Merus thinks it needs a Phase 3 trial to get it past the FDA and to patients.
Clearing the Path for New Uses for Generic Drugs
The labeling-only 505(b)(2) NDA pathway for non-manufacturers to seek FDA approval
Repurposing generic drugs as new treatments for life-threatening diseases is an exciting yet largely overlooked opportunity due to a lack of market-driven incentives. The low profit margins for generic drugs mean that pharmaceutical companies rarely invest in research, regulatory efforts, and marketing for new uses. Nonprofit organizations and other non-commercial non-manufacturers are increasing efforts to repurpose widely available generic drugs and rapidly expand affordable treatment options for patients. However, these non-manufacturers find it difficult to obtain regulatory approval in the U.S. They face significant challenges in using the existing approval pathways, specifically in: 1) providing the FDA with required chemistry, manufacturing, and controls (CMC) data, 2) providing the FDA with product samples, and 3) conducting post-marketing surveillance. Without a straightforward path for approval and updating drug labeling, non-manufacturers have relied on off-label use of repurposed drugs to drive uptake. This practice results in outdated labeling for generics and hinders widespread clinical adoption, limiting patient access to these potentially life-saving treatments.
To encourage greater adoption of generic drugs in clinical practice – that is, to encourage the repurposing of these drugs – the FDA should implement a dedicated regulatory pathway for non-manufacturers to seek approval of new indications for repurposed generic drugs. A potential solution is an extension of the existing 505(b)(2) new drug application (NDA) approval pathway. This extension, the “labeling-only” 505(b)(2) NDA, would be a dedicated pathway for non-manufacturers to seek FDA approval of new indications for well-established small molecule drugs when multiple generic products are already available. The labeling-only 505(b)(2) pathway would be applicable for repurposing drugs for any disease. Creating a regulatory pathway for non-manufacturers would unlock access to innovative therapies and enable the public to benefit from the enormous potential of low-cost generic drugs.
Challenge and Opportunity
The opportunity for generic drug repurposing
On-patent, branded drugs are often unaffordable for Americans. Due to the high cost of care, 42% of patients in the U.S. exhaust their life savings within two years of a cancer diagnosis. Generic drug repurposing – the process of finding new uses for FDA-approved generic drugs – is a major opportunity to quickly create low-cost and accessible treatment options for many diseases. In oncology, hundreds of generic drugs approved for non-cancer uses have been tested as cancer treatments in published preclinical and clinical studies.
The untapped potential for generic drug repurposing in cancer and other diseases is not being realized because of the lack of market incentives. Pharmaceutical companies are primarily focused on de novo drug development to create new molecular and chemical entities. Typically, pharmaceutical companies will invest in repurposing only when the drugs are protected by patents or statutory market exclusivities, or when modification to the drugs can create new patent protection or exclusivities (e.g., through new formulations, dosage forms, or routes of administration). Once patents and exclusivities expire, the introduction of generic drugs creates competition in the marketplace. Generics can be up to 80-85% less expensive than their branded counterparts, driving down overall drug prices. The steep decline in prices means that pharmaceutical companies have little motivation to invest in research and marketing for new uses of off-patent drugs, and this loss of interest often starts in the years preceding generic entry.
In theory, pharmaceutical companies could repurpose generics without changing the drugs and apply for method-of-use patents, which should provide exclusivity for new indications and the potential for higher pricing. However, due to substitution of generic drugs at the pharmacy level, method-of-use patents are of little to no practical value when there are already therapeutically equivalent products on the market. Pharmacists can dispense a generic version instead of the patent-protected drug product, even if the substituted generic does not have the specific indication in its labeling. Currently, nearly all U.S. states permit substitution by the pharmacy, and over a third have regulations that require generic substitution when available.
Nonprofits like Reboot Rx and other non-commercial non-manufacturers are therefore stepping in to advance the use of repurposed generic drugs across many diseases. Non-manufacturers, which do not manufacture or distribute drugs, aim to ensure there is substantial evidence for new indications of generic drugs and then advocate for their clinical use. Regulatory approval would accelerate adoption. However, even with substantial evidence to support regulatory review, non-manufacturers find it difficult or impossible to seek approval for new indications of generic drugs. There is no straightforward pathway to do so within the current U.S. regulatory framework without offering a specific, manufactured version of the drug. This challenge is not unique to the U.S.; recent efforts in the European Union (EU) have sought to address the regulatory gap. In the 2023 EU reform of pharmaceutical legislation, Article 48 is currently under review by the European Parliament as a potential solution to allow nonprofit entities to spearhead submissions for the approval of new indications for authorized medicinal products with the European Medicines Agency. To maximize the patient impact of generic drugs in America, non-manufacturers should be able to drive updates to FDA drug labeling, enabling widespread clinical adoption of repurposed drugs in a formal, predictable, and systematic manner.
The importance of FDA approval
Drugs that are FDA-approved can be prescribed for any indication not listed on the product labeling, often referred to as “off-label use”. Since non-manufacturers face significant challenges pursuing regulatory approval for new indications, they often must rely on advocating for off-label use of repurposed drugs.
While off-label use is widely accepted and helpful for specific circumstances, there are significant advantages to having FDA approval of new drug indications included in labeling. FDA drug labeling is intended to contain up-to-date information about drug products and ensures that the necessary conditions of use (including dosing, warnings, and precautions) are communicated for the specific indications. It is the primary authoritative source for making informed treatment decisions and is heavily valued by the medical community. Approval may increase the likelihood of uptake by clinical guidelines, pathways systems, and healthcare payers. Indications with FDA approval may generate greater awareness of the treatment options, leading to a broader and more rapid impact on clinical practice.
Clinical practice guidelines are often the leading authority for prescribers and patients regarding off-label use. In oncology, for example, the National Comprehensive Cancer Network (NCCN) Guidelines are commonly used guidelines that include many off-label uses. However, guidelines do not exist for every disease and medical specialty, which can make it more difficult to gain acceptance for off-label uses. The Centers for Medicare and Medicaid Services (CMS) policy routinely covers off-label drug uses if they are listed in certain compendia. The NCCN Compendia, which is based on the NCCN Guidelines, is the only accepted compendium that is disease-specific.
Off-label use requires more effort from individual prescribers and patients to independently evaluate new drug data, thereby slowing uptake of the treatments. This can be especially difficult for community-based physicians, who need to remain up-to-date on new treatment options across many diseases. Off-label prescribing can also introduce medico-legal risks, such as malpractice. These burdens and risks limit off-label prescribing, even when there is supportive evidence for the new uses.
As new uses for generic drugs are discovered, it is crucial to update the labeling to ensure alignment with current clinical practice. Outdated generic drug labeling means that prescribers and patients may not have access to all the necessary information to understand the full risk-benefit profile. Americans deserve to have access to all effective treatment options – especially low-cost and widely available generic drugs that could help mitigate the financial toxicity and health inequities faced by many patients. For the public benefit, the FDA should support approaches that remove regulatory barriers for non-manufacturers and modernize drug labeling.
Existing pathways for manufacturers to obtain FDA approval
The current FDA approval system is based on the idea that sponsors have discrete physical drug products. Traditionally, sponsors seeking FDA approval are pharmaceutical companies or drug manufacturers that intend to produce (or contract for production), distribute, and sell the finished drug product. For the purposes of FDA regulation, “drug” refers to a substance intended for use in the treatment or prevention of disease; “drug product” is the final dosage form that contains a drug substance and inactive ingredients made and sold by a specific manufacturer. One drug can be present on the market in multiple drug products. In the current regulatory framework, drug products are approved through one of the following:
- 505(b)(1) NDA: New drug products, including new molecular entities, are first submitted for FDA review through the 505(b)(1) new drug application (NDA) pathway. In this pathway, the sponsor either generates all necessary clinical data or owns the right of reference to the clinical data needed to support the NDA. FDA approval of an NDA establishes a reference listed drug (RLD), based on the FDA’s determinations of safety and effectiveness for the drug.
- 505(b)(2) NDA: For new drug products or formulations utilizing or related to previously approved drugs or drug substances, the 505(b)(2) NDA pathway offers a streamlined alternative to the 505(b)(1) NDA. By allowing the sponsor to reference the FDA’s findings of safety and effectiveness for previously approved drugs and published literature, the sponsor can submit data they do not own or for which they do not have the right of reference, once any exclusivities expire.
- ANDA: Generic drug equivalents are approved through the 505(j) abbreviated NDA (ANDA) pathway. The RLD serves as the standard that other manufacturers may reference when seeking approval for generic versions of the drug. Upon approval, ANDA products receive a therapeutic equivalence designation in the FDA’s Orange Book, indicating the generic drug products are interchangeable for and substitutable with the RLD.
- BLA: Biological products are approved through the biologics license application (BLA). There is currently no provision analogous to section 505(b)(2) under the law governing the approval of biological products. While the FDA is authorized to approve biosimilars, including interchangeable biosimilars akin to generics, no current pathway permits the addition of new indications for use to a biosimilar product short of submitting a complete, original BLA with full preclinical and clinical data. Our proposed regulatory pathway is relevant for small molecule drugs and would not be applicable to biological products under current law.
Manufacturers can add new indications to their approved labeling without modifying the drug product through existing pathways. With supportive clinical evidence for the new indication, the NDA holder can file a supplemental NDA (sNDA), while an ANDA holder may submit a 505(b)(2) NDA as a supplement to their existing ANDA. As previously discussed, the drug product will likely be subject to pharmacy-level substitution with any available therapeutically equivalent generic. The marketing exclusivities that sponsors may receive from the FDA do not protect against this substitution. Therefore, these pathways are rarely, if ever, used by pharmaceutical companies when there are already multiple generic manufacturers of the product.
Challenges for non-manufacturers in using existing pathways
Since manufacturers are not incentivized to seek regulatory approval for new indications, labeling changes are more likely to happen if driven by non-manufacturers. Yet non-manufacturers face significant challenges in utilizing the existing regulatory pathways. Sponsors must submit the following information for all NDAs for the FDA’s review: 1) clinical and nonclinical data on the safety and effectiveness of the drug for the proposed indication; 2) the proposed labeling; and 3) chemistry, manufacturing, and controls (CMC) data describing the methods of manufacturing and the controls to maintain the drug product’s quality.
To submit and maintain NDAs, non-manufacturer sponsors would need to address the following challenges:
- Providing the FDA with required CMC data. NDA sponsors must provide CMC data for FDA review. Non-manufacturers would not produce physical drug products, and therefore they would not have information on the manufacturing process.
- Providing the FDA with product samples. If requested, NDA sponsors must have the drug products and other samples (e.g., drug substances or reference standards) available to support the FDA review process and must make available for inspection the facilities where the drug substances and drug products are manufactured. Non-manufacturers would not have physical drug products to provide as samples, the capabilities to produce them, or access to the facilities where they are made.
- Conducting post-marketing surveillance. Post-marketing responsibilities to maintain an NDA include conducting annual safety reporting and maintaining a toll-free number for the public to call with questions or concerns. Non-manufacturers, such as small nonprofits, may not have the bandwidth or resources to meet these requirements.
Within the current statutory framework, a non-manufacturer could sponsor a 505(b)(2) NDA to obtain approval of a new indication by partnering with a current manufacturer of the drug – either an NDA or ANDA holder. The manufacturer would help meet the technical requirements of the 505(b)(2) application that the non-manufacturer could not fulfill independently. Through this partnership, the non-manufacturer would acquire the CMC data and physical drug product samples from the manufacturer and rely on the manufacturer’s facilities to fulfill FDA inspection and quality requirements.
Once approved, the 505(b)(2) NDA would create a new drug product with indication-specific labeling, even though the product would be identical to an existing product under a previous NDA or ANDA. The 505(b)(2) NDA would then be tied to the specific manufacturer due to the use of their CMC data, and that manufacturer would be responsible for producing and distributing the drug product for the new indication.
As a practical matter, this pathway is rarely attainable. Manufacturers of marketed drug products, particularly generic drug manufacturers, lack the incentives needed to partner with non-manufacturers. Manufacturers may not want to provide their CMC data or samples because it may prompt FDA inspection of their facilities, require an update to their CMC information, or open the door to product liability risks. The existing incentive structure strongly discourages generic drug manufacturers from expending any additional resources on researching new uses or making any changes to their product labeling that would deviate from the original RLD product.
Plan of Action
To modernize drug labeling and enhance clinical adoption of generic drugs, the FDA should implement a dedicated regulatory pathway for non-manufacturers to seek approval of new indications for repurposed generic drugs. Ultimately, such a pathway would enable drug repurposing and be a crucial step toward equitable healthcare access for Americans. We propose a potential solution – a “labeling-only” 505(b)(2) NDA – as an extension of the existing 505(b)(2) approval pathway.
Overview of the proposed labeling-only 505(b)(2) NDA pathway
The labeling-only 505(b)(2) NDA would enable non-manufacturers to reference CMC information from previous FDA determinations and, when necessary, provide the FDA with samples of commercially available drug products. Through this approach, the new indication would not be tied to a specific drug product made by one manufacturer. There is no inherent necessity for a new indication of a generic drug to be exclusively linked to a single manufacturer or drug product when the FDA has already approved multiple therapeutically equivalent generic drugs. Any of these interchangeable drug products would be considered equally safe and effective for the new indication, and patients could receive any of these drug products due to pharmacy-level substitution.
We describe non-manufacturer repurposing sponsors as entities that intend to submit or reference clinical data through a labeling-only 505(b)(2) NDA. This pathway is designed to expand the FDA-approved labeling of generic drugs for new indications, including those that may already be considered the standard of care. Non-manufacturers do not have the means to independently produce or distribute drug products. Instead, they intend to show that there is substantial evidence to support the new use through FDA approval, and then advocate for the indication in clinical practice. This evidence may be based on their research or research performed by other entities, including clinical trials and real-world data analyses.
The labeling-only 505(b)(2) NDA pathway helps address the three major challenges non-manufacturers face in pursuing regulatory approval. Through this pathway, non-manufacturers would be able to:
- Reference the FDA’s previous determinations on CMC data. Currently, a 505(b)(2) NDA can reference the FDA’s previous determinations of safety and effectiveness for an approved drug product. For eligible generic drugs, the labeling-only 505(b)(2) NDA would build on this practice by allowing non-manufacturer sponsors to reference the FDA’s previous determinations on any NDA or ANDA that the manufacturing process and CMC data are adequate to meet regulatory standards.
- Provide the FDA with product samples using commercially available drug product samples. Currently, it is up to the discretion of the FDA whether or not to request samples in the review of an application. With the labeling-only 505(b)(2) NDA, non-manufacturers would provide the FDA with samples of commercially available products from generic manufacturers. Given that the FDA would have already evaluated the products and their bioequivalence to the RLD during the previous reviews, it is not expected that the FDA would need to re-examine the product at the level of requesting samples, except potentially to examine the packaging and physical presentation of the product for compatibility with the new indication and conditions of use. The facilities where the drugs are made would remain available for inspection, under the same terms and conditions as the existing, approved marketing applications.
- Manage post-marketing responsibilities. Since most post-marketing surveillance and adverse event reporting are drug product-specific, these obligations would continue to be the responsibility of the manufacturer of the physical drug product dispensed. With the labeling-only 505(b)(2) NDA, the non-manufacturer would not have product-specific obligations because they are not putting a new product into the marketplace. However, we anticipate the non-manufacturer would be responsible for the repurposed indication on their labeling, including but not limited to post-marketing surveillance as well as indication-specific adverse event reporting and reasonable follow-up.
Under the labeling-only 505(b)(2) NDA, the non-manufacturer sponsor would not introduce a new physical drug product into the market. The new labeling created by the approval would not expressly be associated with one specific product. The non-manufacturer’s labeling would refer to the drug by its established generic name. In that way, the non-manufacturer sponsor’s approval and labeling could be applicable to all equivalent versions of the drug product and would be available for patients to receive from their pharmacy in the same way that generic drugs are typically dispensed at the pharmacy. That is, with the benefit of pharmacy-level substitution, patients could receive any available, therapeutically equivalent drug products from any current manufacturer.
Eligibility criteria
We envision the users of this pathway to be non-manufacturers that conduct drug repurposing research for the public benefit, including organizations like nonprofits and patient advocacy groups. The FDA should implement and enforce additional guardrails on eligibility to ensure that sponsors operate in good faith and cannot otherwise meet traditional NDA requirements. This process may include pre-submission meetings and reviews. The labeling-only 505(b)(2) NDA should be held to the FDA’s standard level of rigor and scrutiny of safety and effectiveness for the proposed indication during the review process.
The labeling-only 505(b)(2) would only be suitable for well-established, commercially available small molecule generic drugs, which can be identified as:
- Drugs with a U.S. Pharmacopeia and National Formulary (USP-NF) monograph. The USP-NF monograph system ensures the uniformity of available products on the market by setting a consensus minimum standard of identity, strength, quality, and purity among all marketed versions of a drug. It is expressly recognized in the Federal Food, Drug, and Cosmetics Act (FDCA). The USP-NF strives to have substance and product monographs for all FDA-approved drugs. USP-NF monographs for generics are commonly available because the drugs have been on the market for a long time and are typically produced by multiple manufacturers. Drug products in the U.S. market must conform to the standards in the USP-NF, when available, to avoid possible charges of adulteration and misbranding.
By statute and regulation, the FDA already allows for NDAs and ANDAs to reference the USP-NF to satisfy some CMC requirements, such as for specifications of the drug substance. As an illustration of the acceptance of the USP-NF, clinical trial protocols requiring the use of background therapy or supportive care, as well as trials testing medical devices requiring the use of a drug product, often will specify that any available version of the drug product meeting USP-NF standards can be used. We propose that products without USP-NF monographs, including certain newer drugs and drugs with especially complicated manufacturing processes that are not conducive to standardization, would not be eligible for the labeling-only 505(b)(2) pathway.
- Drugs with multiple A-rated, therapeutically equivalent products in the FDA Orange Book. The FDA does not regulate which specific products are dispensed or substituted for a given drug prescription. The listing of therapeutic equivalents in the Orange Book facilitates the seamless replacement of drug products from different manufacturers in clinical practice. Therapeutically equivalent drug products: i) have demonstrated bioequivalence to the RLD; ii) have the same strength, dosage form, and route of administration as the RLD; and iii) are labeled for the same conditions of use as the RLD. Therapeutic equivalents that meet these criteria are designated “A-rated” in the Orange Book. A-rated drug products are substitutable for any other version of that A-rated drug product, including the RLD itself.
Implementation
The labeling-only 505(b)(2) NDA pathway could be implemented through an FDA guidance document interpreting the current statute and regulations or through legislation that clarifies the FDA’s existing authority. Guidance documents contain the FDA’s interpretation of a given policy on regulatory issues. The FDA’s Center for Drug Evaluation and Research (CDER) could issue new guidance that allows for interpretation of the existing statute, thereby officially authorizing previous FDA determinations of acceptable CMC data to be referenceable for eligible generic drugs and adjusting drug sample requirements. Alternatively, the labeling-only 505(b)(2) could be enacted by Congress through a statutory change by incorporation into FDCA, which is up for reauthorization through the Prescription Drug User Fee Act (PDUFA) in 2027, or other congressional acts as appropriate. FDA guidance would be a faster pathway to adoption, while statutory authorization would offer additional safeguards for the continuance of the pathway long term.
The labeling-only 505(b)(2) NDA pathway could be funded through user fees, which are established by PDUFA for the cost to file and maintain NDAs. However, many nonprofit sponsors would not be able to afford the same user fees as for-profit pharmaceutical manufacturers. Relevant statutes will likely need to be updated to create a different fee schema for non-manufacturers who use the labeling-only 505(b)(2) pathway. In a similar spirit to reducing barriers to maintaining up-to-date labeling, the 2017 PDUFA update waived the fee for submitting an sNDA, which is how an existing RLD holder would update their labeling with new indication information.
Conclusion
Patients need new and affordable treatment options for diseases that have a devastating societal impact, and repurposing generic drugs can help address this need. Nonprofits and other non-manufacturers are driving these efforts forward due to a lack of interest from pharmaceutical companies. As momentum gains for generic drug repurposing, the U.S. regulatory system needs a pathway for non-manufacturers to seek FDA approval of new indications for existing generic drugs. Our proposed labeling-only 505(b)(2) NDA would eliminate undue administrative burden, enabling non-manufacturers to pursue FDA approval of new indications. It would allow the FDA to provide the public with the most up-to-date drug labeling, improving the ability of patients and physicians to make informed treatment decisions. This dedicated pathway would increase the availability of effective treatment options while reducing costs for the American healthcare system.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
The FDA does not have sufficient bandwidth or resources to meet the opportunity we have with repurposed generic drugs. To be the primary driver of labeling changes for repurposed generic drugs, the FDA would need to identify repurposing opportunities and also thoroughly compile and evaluate the safety and effectiveness data for the new indications. Project Renewal in FDA’s Oncology Center of Excellence is working with RLD holders to update the labeling of certain older oncology drugs where the outdated labeling does not reflect their current clinical use. The initial focus of Project Renewal is limited, and newly repurposed treatments are not included within its scope. For newly repurposed treatments, the FDA could evaluate the drugs and post to the Federal Register reports on their safety and effectiveness that could be referenced by manufacturers. However, this approach is burdensome as it would require a significant commitment of FDA resources. By introducing a motivated, third-party non-manufacturer as the primary driver for labeling changes, non-manufacturers can contribute expertise and resources to enable faster data evaluation for more drugs.
The pre-existing NDA sponsor could update their labeling to add the new indication through an sNDA that references the labeling-only 505(b)(2) NDA. ANDA holders would then be legally required to match their labeling to that of the RLD. The FDA should determine whether all current manufacturers would be required to update their labeling following approval of the new indication, and if so, the appropriate process.
Generic drugs play a vital role in the U.S. healthcare system by decreasing drug spending and increasing the accessibility of essential medicines. Generics account for 91% of prescriptions filled in the U.S. Expanding the market with generics for new indications could lead to short-term price increases above the inflation rate for off-patent branded and generic drugs in some unlikely circumstances. For example, if a new use for a generic drug substantially increases demand for the drug, there is a short-term risk that prices for the drug or potential substitutes could rise until manufacturers build more capacity to increase supply. To mitigate this risk, generic manufacturers could be notified about potential increases in demand so they can plan for increased production.
Generally, healthcare payers are not required to cover or reimburse for off-label uses of drugs. Unless a drug undergoes utilization management, payers cover most generic drugs for off-label uses because coverage is agnostic of indication. Clinical practice guidelines are highly influential in the widespread adoption of off-label treatments into the standard of care and are often referenced by payers making reimbursement decisions. In oncology, many off-label treatments are included in guidelines; only 62% of treatments in the NCCN are aligned with FDA-approved indications. For example, more than half of the NCCN recommendations for metastatic breast cancer are off-label treatments. Due to the breadth of off-label use, we anticipate payers would continue to cover repurposed generic drugs used off-label, even if there is a pathway available for non-manufacturers to pursue FDA approval.
We do not envision any form of exclusivity being granted for indications pursued via a labeling-only 505(b)(2) NDA. Given that the non-manufacturer sponsor would rely on existing products produced by multiple generic manufacturers, there is no new product to grant exclusivity. Even if some form of exclusivity were given to the non-manufacturer, it would be insufficient to guarantee the use of any particular drug product over another due to pharmacy-level substitution.
Advisory Committees for the 21st Century Recommendations Toolkit
From January 2024 to July 2024, the Federation of American Scientists interviewed 30 current and former Advisory Committee (AdComm) members. Based on these discussions, we were able to source potential policy recommendations for the executive level, for the executive level, that may assist with enhancing the FDA’s ability to obtain valuable advice for evidence-based decision-making. In this toolkit, we build off of those discussions by providing you with actionable policy reform recommendations. We hope that these recommendations catapult the Advisory Committee structure into one best suited to equip all AdComms with the necessary tools needed to continue providing the government with the best advice.
Download a PDF version of these recommendations on the left.
Recommendations and Best Practices for Actionable Policy Implementation
Voting for FDA Advisory Committees
Update Type: Process
- Maintain voting as an integral function, allowing FDA Advisory Committee members to convey their collective expertise and advice, aiding the FDA in informed decision-making on scientific and regulatory matters.
- Revise the guidance for FDA Advisory Committee Members and FDA Staff to explicitly define circumstances for which voting should occur and eliminate sequential voting
Best Practices for Implementation
The United States Food and Drug Administration can uphold their voting mechanism by updating their document entitled, “Guidance for FDA Advisory Committee members and FDA staff: Voting Procedures for Advisory Committee Meetings” to include language that clearly states a vote should be taken at all Advisory Committee meetings where a medical product is being reviewed. This guidance should also indicate that the absence of voting should only occur if an Advisory Committee meeting has been convened to discuss issues of policy. Further, this guidance should be considered a level 2 guidance as it falls into the category of addressing a “controversial issue”. To effectuate these changes, a notice of availability (NOA) may be submitted to the Federal Register for public input (public input is not a requirement before implementation).
Potential Language to be Utilized for Guidance
In an effort to continue to allow Advisory Committee members to provide unbiased, evidence-based feedback and uphold such an integral part of the Advisory Committee process, voting is hereby mandatory for all Advisory Committee meetings that are convened where the purpose is to review and assess the safety and efficacy of medical products.
Involved Stakeholders
In order for this process to successfully occur, the FDA will need to amend their guidance with the aforementioned updates. Consideration should be given to incurred costs for personnel required to complete these updates. Personnel needed for these amendments may include, but are not limited to, the (a) Office of the Commissioner, Office of Clinical Policy and Programs, Office of Clinical Policy, (b) Center for Devices and Radiological Health, (c) Center for Biologics Evaluation and Research, and (d) Center for Drug Evaluation and Research.
FDA Staff, Leadership, and AdComm Disagreements
Update Type: Process
- Ensure that all FDA staff and leadership are fully cognizant of the existence and details of the Scientific Dispute Resolution at FDA guidance and the process for submitting disputes for review.
- Develop guidance that clearly explains a transparent process to communicate effectively with AdComm members regarding decision making when parties have opposing viewpoints
Best Practices for Implementation
Implementing these recommendations will improve conflict resolution internally and between the Agency and Advisory Committee members. Best practices for implementation include (a) building the Scientific Dispute Resolution at FDA guidance into the official FDA onboarding process for new hires to raise awareness, (b) provide annual employee trainings in an effort to stay up-to-date with dispute resolution processes and procedures, and (c) develop a guidance that delineates the process for resolving conflicts between the Agency and Advisory Committees when there are differing opinions.
Note: Guidance for resolving disputes between the Agency and Advisory Committees should be submitted to the Federal Register for public comment. Guidance should also include plain language that designates the avenue to be used for official decision notifications, the timeliness of these notifications after convenings have concluded, and circumstances in which the FDA cannot notify Advisory Committees that their decision is in direct opposition of the Committee’s vote (e.g., – if this notification would breach a confidentiality agreement with the applicant). Implementing a transparent process to communicate with AdComm members regarding differences between the Agency and the AdComm will assist in improving morale between both parties, but also encourage continued support of the AdComm.
Involved Stakeholders
Successful implementation of these recommendations will require the capacity of human resources personnel and individual center leadership.
Update Type: Regulatory
- Incorporate the Scientific Dispute Resolution at FDA guidance into FDA regulations
- Amend the Scientific Dispute Resolution at FDA guidance to dictate the mandatory execution of best practices within the dispute resolution process
- This guidance should identify additional non-biased parties (that may not be government-affiliated) to provide impartial guidance on complex scientific matters affecting public safety.
Best Practices for Implementation
Center leadership can assign FDA staff to make the necessary guidance amendments which should include the requirements for inclusion in the onboarding of all FDA employees. Staff should also be responsible for obtaining feedback on amendments from all necessary internal parties and submitting proposed amendments to OMB for review and addition to the Federal Register. Federal register comments will then be reviewed by FDA staff, guidance will be updated accordingly, and a final draft submitted to OMB for review. If approved, the final regulation will be published in the Code of Federal Regulations. Congressional involvement should not be necessary.
FDA Center leadership should delegate the task of creating an annual mandatory training program for all FDA employees to review this guidance in an effort to stay abreast of the procedures for dispute resolution.
Involved Stakeholders
FDA Center leadership, FDA staff, and the Office of Management and Budget (OMB) are the intended stakeholders for implementation of these recommendations. To incorporate this guidance into FDA regulations, Center leadership will need to assign FDA staff to amend guidance and submit to OMB.
Leveraging AdComm Membership
Update Type: Process
Expanding Committee Representation
- If there is flexibility and committee composition is not bound by law, include a patient representative and pharmacist and/or pharmacologist on each Committee
- Patient representatives provide a needed perspective due to lived experiences and understanding of how specific drugs and devices affect their day-to-day life
- Drugs and devices will usually pass through the hands of pharmacists and pharmacologists. Therefore, they should have the opportunity to serve on these Committees and provide feedback. Pharmacologists also understand the clinical application of drugs.
- Have a roster of temporary members that can be used when additional expertise is needed for Committee meetings or when there is a conflict of interest (COI)
Amplifying the Role of the Chair
- Expand the role of the Committee chair that will encompass the task of recruiting both standing and ad hoc members, as well as identifying prominent issues and products for Committee consideration, thereby allowing for specialized input from their Committee
Establishing Training and Regulatory Procedures for Incoming Members
- Institute a basic 101 training for all newly appointed Advisory Committee members that covers statistical analysis, clinical trial design, and elucidates the partnership between the FDA and AdComm
- Include an overview of the regulatory process and how the FDA’s decision-making process is performed
Best Practices for Implementation
With respect to Committee composition, the FDA should consider adding patient representatives to all Committees that review medical products. The addition of a patient representative will ensure that the voice of the population who the medical product affects is heard. The FDA can select individuals best suited to fill these roles through connecting with patient advocacy organizations. If patient representatives are selected, the FDA should develop an onboarding program to familiarize the patient representative with basic knowledge of the federal regulation process. This program should educate the patient representative on (a) the types of questions presented to Advisory Committees, (b) how the FDA views the role of the patient representative in the process, (c) the internal review process for data that is submitted, and (d) other pertinent topics related to medical product regulation.
Leveraging the role of the Advisory Committee Chair can help the FDA fully optimize the use of their Committee. “Chairs possess extensive networks that could support the identification of permanent or temporary expert participants for AdComms” (Banks, 2024). Allowing Chairs the ability to identify relevant issues or products for their respective committees to review can provide an additional layer for the FDA to keep abreast of critical public concerns via appropriate committee evaluation (Banks, 2024).
Finally, while Committee members may be experts in their own right, training should be provided for all. The FDA should provide basic 101 training courses that can cater to the needs of members with various knowledge bases. Training should include information on the relationship between the FDA and Advisory Committee members, best practices for understanding statistical analysis, and the different types of clinical trial designs. Training should provide real-world examples of statistical analysis and trial design in use (this can be done by providing examples from prior medical product review).
Involved Stakeholders
FDA Center staff (including statisticians and scientists for the development of training programs).
Conflict of Interest (COI) Auditing
UPDATE TYPE: PROCESS
- Develop a process that can quickly replace individuals who have a known conflict of interest
- Clearly delineate criteria for committee service acceptance regarding individuals with potential or actualconflicts of interest
Best Practices for Implementation
To prevent recurring COIs, the FDA should develop a database of experts for various categories of expertise that can be selected to replace those with known COIs. This database should include names, contact information, credentials, all areas of expertise for each expert, and should link to public financial interest databases that can serve as a source for identifying conflicts (e.g., Open Payments, Dollars for Docs).
To prevent public confusion, the FDA should amend their Guidance for the Public, FDA Advisory Committee Members, and FDA Staff: Public Availability of Advisory Committee Members’ Financial Interest Information and Waivers to specify circumstances that warrant a COI waiver being administered. This will increase transparency and help Advisory Committee members and the public understand the reasoning in allowing members with conflicts to participate in meetings. This guidance can then be submitted to the Federal Register for public comment
UPDATE TYPE: PROCESS | REGULATORY
- Streamline the COI process to prevent duplicative work that may act as a deterrent to experts volunteering to serve on the AdComm (work with GSA on this matter if necessary)
Best Practices for Implementation
Streamlining the COI process will assist the FDA with retention efforts for Advisory Committees while maintaining compliance with conflict of interest regulations. A digital system should be developed that allows Advisory Committee members to select whether their financial information has changed through the use of a dropdown or check box. This will prevent duplicative work and also contribute to a sustainable (green) process.
Involved Stakeholders
The Office of the Commissioner, Office of Clinical Policy and Programs, Office of Clinical Policy would be the interested stakeholder to issue updates to the COI policy and would work with the General Services Administration if necessary.
The Role of Patient Advocacy in the AdComm Process
Update Type: Process
- Dedicate staff to identifying crucial public comments from patient advocates that should be considered for regulatory decision-making
- Include a patient representative on all committees that are reviewing medical products
- If possible, ensure that patient representatives selected have basic knowledge of the federal regulation process
- Establish criteria for which all public comments must abide by
- This can be done by creating a checklist for the public to review and consider before forming their comment and that clearly delineates the mandatory criteria that a medical product must meet to be considered for approval
- A disclaimer can also be added to state that there is a specific threshold or sample size of a population who must benefit from the medical product
- Promote patient focused medical product development through the use of incorporating patient perspectives into the life cycle of the regulatory process
- This can be done through hosting patient town hall discussions for areas such as rare diseases and expanding initiatives such as the Patient Focused Drug Development (PFDD) public meetings
- Make patient engagement ongoing, rather than only allowing patient engagement quarterly or annually as done with most FDA-led programs and initiatives
Best Practices for Implementation
Public comments are a crucial part of the regulatory process. The FDA should focus on increasing participation in this process, as well as notifying participants of what should be expected from the public comment period. By law, the FDA must allow a public comment period for all Advisory Committee convenings. To increase participation, the FDA should allow public comments to be vocalized in-person and virtually, in addition to the submission of public comments to the docket.
Regarding Committee composition, please refer to Best Practices for Implementation under Leveraging AdComm Membership for information on the addition of patient representatives to Advisory Committees (see prior section above).
Patient engagement in the regulatory process is necessary to inform evidence-based decisions. While the FDA currently has ways to engage these communities through the use of public comment and initiatives such as Patient Focused Drug Development (PFDD), engagement should be an ongoing process. As mentioned, the FDA can develop and leverage existing relationships with public health agencies and advocacy organizations who can then serve as the liaison of feedback to the FDA. The FDA can also consider expanding their current initiatives and programs to engage communities twice a quarter instead of quarterly or annually. Consistent engagement in this form will help to establish trust between the FDA and the public who they serve, as well as give them the needed information from the communities who are most impacted from their decisions.
Involved Stakeholders
Implementation of these recommendations and expansion of current initiatives will require the involvement of FDA Center leadership and Center staff.
Improving Public Awareness and Understanding of Advisory Committees
Update Type: Process
- FDA can leverage social media platforms to increase awareness and understanding of AdComms through the use of disseminating information (engagement, ads, etc.)
- FDA should include a disclaimer on all communications and marketing materials regarding AdComms
- These disclaimers should also be made at all public meetings
- Disclaimers should emphasize the purpose of AdComm votes (disclaimer should state that votes allow AdComm members to provide an official stance to the FDA as experts, but those votes are also non-binding)
- Develop a webpage that allows people to be placed on listserv regarding upcoming meetings
- Partner w/state and local public health agencies and advocacy organizations to spread awareness
Best Practices for Implementation
The FDA should develop a monthly content plan to utilize its current interactive and social media outlets and disseminate information related to the role of Advisory Committees and their convenings, while also maintaining compliance with the FDA’s social media policy. The social media content plan should be centered around (a) what an Advisory Committee is, (b) how members are selected, (c) information regarding votes of Advisory Committees and how they are specific to safety and efficacy, but are not voting on the approval of a medical product, (d) discussing upcoming Advisory Committee meetings, their location, and inviting the public to participate via public comment, (e) sharing information about what a public comment is, requirements for making public comments, and how the FDA reviews them, and (f) sharing a webpage where the general public can input their personal email to be notified of upcoming Advisory Committee meetings.
FDA staff should develop a plain language disclaimer to be placed on all social media posts, meeting materials, and websites related to Advisory Committees. This disclaimer should illustrate that Advisory Committee members will provide unbiased expertise to assist the FDA with their decision. However, while the Committee’s vote is included in consideration for the decision, their vote is non-binding which leaves the FDA as the final decision maker for approval.
Finally, the FDA should identify state/local public health agencies, as well as advocacy organizations that they can potentially partner with to disseminate information more broadly. These agencies and advocacy organizations have strong relationships with various communities who should be engaged in the regulatory process. Developing a relationship with these agencies and organizations in an attempt to engage the community will assist the FDA with building connections and trust, as well as mutual understanding of Advisory Committee roles. Potential partners can be identified using the linked list under involved stakeholders.
Involved Stakeholders
Implementation of these recommendations and expansion of current initiatives will require the involvement of FDA Center leadership, Center staff, Office of External Affairs (OEA) Web and Digital Media staff, Office of Information Management and Technology (OIMT), state/local public health departments, and advocacy organizations.
Note: These listings of state/local public health departments and advocacy organizations are intended to be used as a starting point in the identification of potential partners and not to be considered an exhaustive list.
For questions related to this toolkit, please contact
Cheri Banks
Health Regulatory Specialist
cbanks@fas.org
Grace Wickerson
Health Equity Policy Manager
gwickerson@fas.org
Improving Public Awareness and Understanding of Advisory Committees
From January 2024 to July 2024, the Federation of American Scientists interviewed 30 current and former Advisory Committee (AdComm) members. Based on these discussions, we were able to source potential policy recommendations that may assist with enhancing the FDA’s ability to obtain valuable advice for evidence-based decision-making. The results of these discussions are presented in case study format detailing the recurring themes that emerged and policy recommendations for improvement.
The FDA holds one of the most important roles as a federal agency which is to ensure public safety when approving vaccines, medical devices, and medicines. The approval of these products usually require extensive trials with data that supports their safety and efficacy. Considering that most of these decisions are complex and multifaceted, the FDA enlists the support of Advisory Committees to assist with their decision-making process. The primary role of FDA Advisory Committee members is to provide the FDA with informed advice and recommendations on issues spanning science, regulatory policy, and the evaluation of products under the FDA’s jurisdiction. Although AdComm members serve the FDA in an advisory capacity, their recommendations are non-binding. Therefore, they do not have the final say in the regulatory approval process.
However, over the years, it has been made evident that the public is unaware of the role of Advisory Committees and ways in which they can engage with the FDA. In this case study, FAS hopes to share the current problem and actionable recommendations to combat public misconceptions regarding FDA AdComm roles and provide guidance on increasing FDA engagement with the public and other relevant stakeholders throughout the regulatory process.
Public Awareness Problems
While AdComm members are experts in their respective fields and volunteer their time to provide advice to the FDA, there are multiple factors that must be considered before making official decisions. The recommendations provided during Advisory Committee meetings are just one aspect that is considered for regulatory decision-making and do not guarantee an official approval or denial of a product by the FDA. During AdComm meetings, the FDA allows the general public to make public comments to the Agency and the AdComm regarding the topic that is being addressed. Despite this, members of the general public have expressed that, on many occasions, they are unaware AdComm meetings are occurring. This, in effect, deprives them of the opportunity to communicate directly with the FDA and the AdComm. Additionally, they feel the FDA fails to engage them in an adequate manner, thereby limiting opportunities for participatory engagement. It has also been noted that most members of the general public are unaware FDA Advisory Committees exist; and, for those who are aware, they are unclear about the capacity of their role within the regulatory process.
For these reasons, the FDA must take measures to enhance public understanding in an effort to combat misinformation, educate, and raise awareness on the existence of Committees and their purpose.
Communicating AdComms to the Public
Improving Public Awareness of Advisory Committees and their Role
Improving public awareness on the existence of FDA Advisory Committees and their purpose would assist the FDA with improving public trust and debunking myths and misinformation related to the approval of medical products. Advisory committees operate as an independent party and their recommendations assist with guiding regulatory decision-making. However, their recommendations are non-binding, and FDA leadership must consider additional factors before granting approvals or denials of medical products.
To increase public awareness on Advisory Committees, it should be made clear that AdComm recommendations are not conclusive, as the FDA considers multiple factors in its official decisions. The FDA can leverage social media platforms to increase awareness and understanding of AdComms through the use of disseminating information via the use of ads and active social media engagement. A survey conducted by Pew Research Center states that eight in ten Americans believe social media platforms are an effective way to bring awareness. In addition, disclaimers should be included on all public facing materials referencing AdComms to indicate their purpose. Clearly communicating this to the public will dispel myths that AdComms make the final call on the approvals of medical products.
Improving Communication about Advisory Committee Meetings
Encouraging public participation for Advisory Committee meetings will help foster a collaborative and engaged general public who can contribute valuable life experience to the regulatory process. FAS has identified ways in which the FDA can better communicate with the public to inform them of Advisory Committee meetings. First, the FDA can develop a webpage that allows people to receive notifications of upcoming AdComm meetings. The FDA can also establish relationships with state and local public health agencies, as well as advocacy organizations to spread awareness. Through these relationships, the various agencies and organizations can use their networks to disseminate widespread information on AdComm meetings. Public health agencies and advocacy organizations can gauge the best ways in which these communities would like the FDA to engage with them. This understanding of the communities they serve makes them an ideal partner for fostering continuous engagement.
Policy Recommendations
In an effort to improve public awareness and understanding of AdComms, the potential policy recommendations are as follows:
- FDA can leverage social media platforms to increase awareness and understanding of AdComms through the use of disseminating information (e.g., engagement, ads, etc.)
- FDA should include a disclaimer on all communications and marketing materials regarding AdComms
- These disclaimers should be made at all public meetings
- Disclaimers should emphasize the purpose of AdComm votes. Disclaimers should state that votes allow AdComm members to provide an official stance to the FDA as experts, but votes are non-binding.
- Develop a webpage that allows people to be placed on a listserv regarding upcoming meetings
- Partner with state and local public health agencies and advocacy organizations to spread awareness
Conclusion
Advisory Committees are essential to the FDA regulatory decision-making process. It’s imperative that their role is understood by them and the general public to best move the needle forward. While the FDA currently allows the public to provide public comment at Committee meetings, that alone cannot be considered engaging the community. The FDA must create new opportunities for interpersonal communication which will create an environment of mutual trust and understanding between both parties.
The Role of Patient Advocacy in the AdComm Process
From January 2024 to July 2024, the Federation of American Scientists interviewed 30 current and former Advisory Committee (AdComm) members. Based on these discussions, we were able to source potential policy recommendations that may assist with enhancing the FDA’s ability to obtain valuable advice for evidence-based decision-making. The results of these discussions are presented in case study format detailing the recurring themes that emerged and policy recommendations for improvement.
The regulation of medical products is the responsibility of the Food and Drug Administration (FDA). To ensure effective decision-making regarding these products, the FDA recognizes the importance of patient advocacy and the perspectives of patients. In 1988, the FDA initiated the patient engagement process through the Office of Aids Coordination, and within five years, the first patient representative was appointed to an FDA Advisory Committee. Since then, the FDA has significantly enhanced its methods of engaging patients, caregivers, and patient advocates. This includes the establishment of various offices, programs, collaboratives, listening sessions, public guidance, and more.
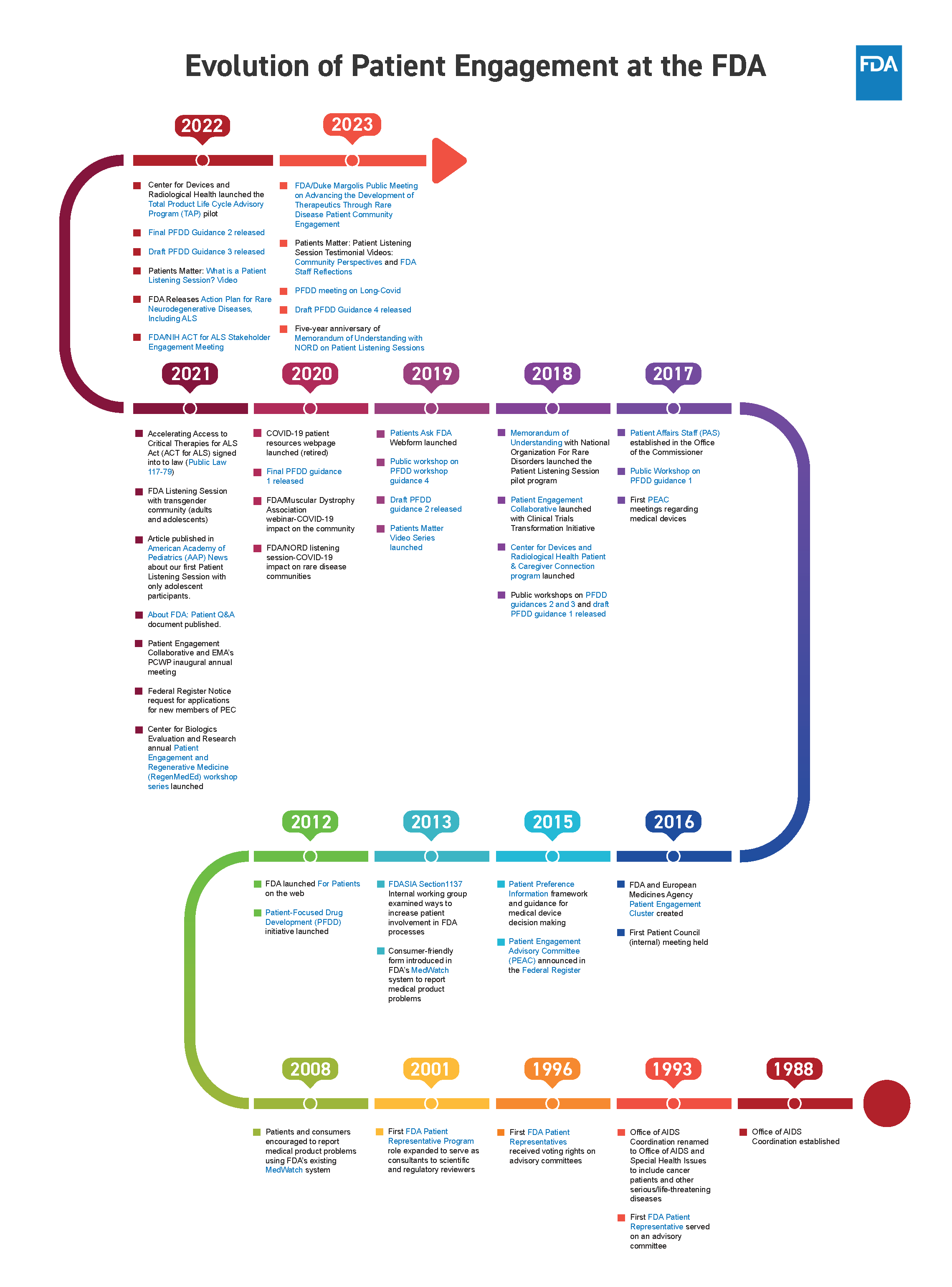
The FDA employs several avenues to engage patients in the regulatory process. Some avenues include the Patient Engagement Advisory Committee (PEAC), public comment, the Patient Focused Drug Development Initiative (PFDD), the Patient Listening Session Program, Patient Engagement Collaborative (PEC), and the Patient Representative Program (PRP). This list does not cover all the ways in which the FDA engages patients and advocates but provides an overview of the key operations involved in patient engagement efforts.
The Patient Engagement Advisory Committee is the only Committee that is completely composed of caregivers, patients, and patient representatives from various organizations, as a way to ensure that the lived experiences of these populations and their opinions are included in the deliberations and regulatory decision-making of medical products. Public comment is a requirement by law for federal agencies, allowing the public to provide feedback on proposed actions or new rules and regulations. Public comment is also sought during Advisory Committee meetings to gather information and perspectives from the public. PFDD meetings provide a platform for the FDA to obtain insights from patients on specific diseases and their treatments. To identify the issues most important to patients, the FDA has a series of guidance documents that are used specifically for PFDD meetings.
The Patient Listening Session Program facilitates informal meetings between patients, their representatives, and FDA staff. These sessions cover a range of topics, including treatment preferences, quality of life, unmet medical needs, and the impact of diseases and their symptoms. The PEC offers a forum for patients, caregivers, and advocates to discuss patient engagement operations. Lastly, the PRP allows patients, caregivers, and advocates who serve as special government employees the opportunity to provide advice to the FDA’s Commissioner or a designated representative on matters related to medical devices and their regulation.
Although there are various avenues for patient engagement and advocacy participation in the medical product regulation process, there are also ways in which these avenues can be expanded or improved.
Patient Advocacy Problems
For many years, patients and their caregivers have not seen significant or sustainable treatments that have been developed to treat many illnesses and diseases. Some treatments have proven to be ineffective yet still made it to market approval. On the other hand, there are treatments that met safety and efficacy standards but were not approved. There are also those treatments that are simply not affordable to the populations that need them most. In many of these scenarios, the patient representative voice was lost as they did not have the option to express their concerns or perspectives on certain treatments with decision makers. This further confirms that the role of patient advocacy and allowing space for the patient representative voice is crucial to the regulation process of medical products.
At the moment, the patient voice is not always heard because there are some FDA Advisory Committees that do not have a patient representative.
While the role of patient advocacy is crucial, it is important to note there should be boundaries in which patient perspective is considered for decision-making. Although patients and their advocates seek treatments that better address their needs, this desire can sometimes obscure their judgment concerning long-term treatment effectiveness. Frequently, patients and their supporters present powerful arguments to Advisory Committees and the FDA for approval of particular medical products which can lead to expedited medical product approval in the absence of supportive evidence.
The endorsement of eteplirsen, which was intended for the treatment of Duchenne muscular dystrophy (DMD), illustrates this point. Despite a 7 to 3 vote by the FDA’s Advisory Committee against approval due to insufficient evidence of its benefits, opposition from the FDA’s Center for Drug Evaluation and Research’s former leader resulted in the drug’s authorization. This sparked substantial internal and public criticism and led Dr. Ellis Unger from the FDA’s Office of Drug Evaluation to challenge the approval decision. Dr. Unger emphasized that “patient-focused drug development is about listening to patient perspectives about what matters to them; it is not about basing drug approvals on anecdotal testimony that is not corroborated by data.”
This approval was perceived by many as having been heavily influenced by patient advocacy and raised concerns about potential long-term implications for patient health. It also signaled a need to further examine both patient education and the appropriate limits of patient involvement in the regulatory process. This could have been mitigated had there been a list of criteria in place to be followed for public comment.
Incorporating Patient Perspectives
The Food and Drug Administration (FDA) is committed to understanding the balance of benefits and risks acceptable to patients as they relate to medical products. The FDA defines the role of patient representatives that serve on Advisory Committees as “Special Government Employees” who provide direct input to agency staff and share valuable insight on their experiences with various diseases, conditions, and devices while gaining access to confidential information. These representatives are selected by the FDA to serve on Advisory Committees using a specific set of criteria including, but not limited to:
- “Personal experience with the disease either as a patient or primary caregiver”
- “Knowledge about most treatment options and research for their areas of experience they are representing”
- “Impartiality and compliance with Federal ethics requirements (for example, financial interest, such as stock, in companies that may be affected by FDA decisions)”
This criteria ensures the FDA will understand the patient perspective as it relates to various medical products and ensures those selected to serve on Advisory Committees are knowledgeable about the areas in which they are aiming to provide guidance. Currently, there are some FDA Advisory Committees that do not have a patient representative. Further, the patient representatives serving on committees do not always have voting privileges. The absence of consistent voting privileges for some patient representatives on Advisory Committees and not having a standing patient representative on all committees hinders these individuals from providing an official stance on behalf of the community they represent. Additionally, public comment plays a significant role at Advisory Committee meetings by permitting individuals—including patients, caregivers, and advocacy organizations—to highlight concerns and propose solutions that may not have been previously considered by decision-makers. This process also helps the committee and agency gauge patient acceptance or opposition related to medical products, thereby enhancing their ability to make decisions that more accurately reflect public needs.
When Sarepta was seeking approval of eteplirsen for the treatment of DMD, a patient advocacy organization brought hundreds of patients, caregivers, and other advocates to the Advisory Committee convening so they could make a public comment to the Committee and the agency. Shortly after, the drug received a swift approval. Although it presented much controversy within the agency and the public, it showed how influential patient advocacy can be. Personal lived experiences, compelling stories of debilitating illnesses, and experiences with current treatment have the ability to impact regulatory decision-making.
The role of patient advocacy continues to be important in the Advisory Committee process and FDA regulatory decision-making process because it is crucial to assisting with decisions that affect the American public. Patient advocacy can be presented in the form of patient representatives that serve on Advisory Committees, those who make public comments during Advisory Committee convenings, and various outreach programs by advocacy organizations. The role of advocacy gives patients and caregivers support, promotes and protects their rights, and allows broader visibility for the issues that are most important to them. All of these avenues for patient perspective are important to understand how treatments perform, the current needs of the patient population, and how to tailor care for these populations by truly understanding their condition, diagnosis, and current management. Therefore, their voice is critical to truly understanding how various medical products will benefit their population, how they will access and afford these products, and how they will fill an unmet medical need.
Policy Recommendations
In an effort to better leverage Advisory Committee membership, the potential policy recommendations are as follows:
- Dedicate staff to identifying crucial public comments from patient advocates that should be considered for regulatory decision-making
- Include a patient representative on all committees that are reviewing medical products
- If possible, ensure that patient representatives selected have basic knowledge of the federal regulation process
- Establish criteria for which all public comments must abide by
- This can be done by creating a checklist for the public to review and consider before forming their comment and that clearly delineates the mandatory criteria that a medical product must meet to be considered for approval
- A disclaimer can also be added to state that there is a specific threshold or sample size of a population who must benefit from the medical product
- Promote patient focused medical product development through the use of incorporating patient perspectives into the life cycle of the regulatory process
- This can be done through hosting patient town hall discussions for areas such as rare diseases and expanding initiatives such as the Patient Focused Drug Development (PFDD) public meetings
- Make patient engagement ongoing, rather than only allowing patient engagement quarterly or annually as done with most FDA-led programs and initiatives
Conclusion
The landscape of disease burden and associated symptoms is ever-evolving. To ensure the FDA is best prepared for this changing landscape, patient advocacy and amplifying the patient voice should be considered vital to the development and regulation of medical products. Involving those who are the most impacted by these products is essential. The FDA can further promote the patient perspective and advance patient-centered health through incorporating patient representatives on all Committees that are reviewing medical products, making patient engagement an ongoing process, hosting town halls for patients to allow a broader audience the opportunity to voice opinions, and having dedicated staff to sort through public comments from patients.
FDA Staff and Leadership Disagreements and the Role of the AdComm in the Regulatory Process
From January 2024 to July 2024, the Federation of American Scientists interviewed 30 current and former Advisory Committee (AdComm) members. Based on these discussions, we were able to source potential policy recommendations that may assist with enhancing the FDA’s ability to obtain valuable advice for evidence-based decision-making. The results of these discussions are presented in case study format detailing the recurring themes that emerged and policy recommendations for improvement.
The FDA relies on its scientific staff and Advisory Committees to provide conclusions from trial and study data, which aid in the process of regulating and approving medical products. However, there are instances when disagreements arise between the agency’s scientists, statisticians, Advisory Committees, and leadership on the accelerated or full approval of medical products. The resolution of these disagreements present a growing concern about FDA leadership overruling the expert opinions of scientific staff and proceeding with official approvals, thus undermining staff expertise, decreasing agency morale, and potentially diminishing public trust.
When Disagreements Between FDA and AdComms Arise
The Federal Advisory Committee Act governs the FDA’s Advisory Committees and establishes a process in which the FDA can seek expert advice on various issues related to science, regulatory policy, and the evaluation of products under the FDA’s jurisdiction. When the FDA has differing opinions on safety and efficacy requirements of medical products, certain products may be referred to an Advisory Committee for further data review by an impartial entity. To aid in this matter, the FDA has developed guidance detailing the process for assembling Advisory Committees. Since the resources required for convening these committees are significant, the FDA ensures there is substantial uncertainty or disagreement regarding the data. Advisory Committees will discuss the evidence and provide feedback with the goal of producing the most optimal evidence-based resolution. This part of the regulatory process is crucial to the agency’s regulatory decision-making as it involves unbiased parties and leads to transparency, upholding public safety, and maintaining public trust. However, there are times that FDA leadership disagrees with the votes of their Committees and proceeds with controversial approvals. One example of this scenario was approval of the drug Aduhelm. Aduhelm, which was marketed as a treatment for Alzheimer’s, received an overwhelmingly negative vote from the Advisory Committee to move forward with its approval for market distribution. Ten of eleven Committee members stated the data did not support adequate efficacy for approval. Nonetheless, the FDA granted accelerated approval, sparking resignations from a third of their Committee and outrage amongst the public. This disregard for expert opinion was viewed as the FDA exhibiting an approval bias, a perspective the public currently maintains of the FDA.
In 2023, various organizations and coalitions, such as Doctors for America, Jacob’s Institute of Women’s Health, National Center for Health Research, TMJ Association and more publicly expressed concerns regarding the FDA’s leadership and their approach to drug approvals through a letter addressed to the FDA’s Chief Scientist, Dr. Namandje N. Bumpus. They highlighted FDA leadership had ignored claims by scientific staff that safety and efficacy standards were not being met for the drug Elevidys. Manufactured by Sarepta Therapeutics, Elevidys was granted accelerated approval by FDA leadership. Dr. Bumpus responded to this letter defending the FDA’s approval of Elevidys by referring to the agency’s comprehensive review of the data and consideration of the potential risks of the approved treatment, nature of the disease and its impact on patients, and the limited amount of therapies available. However, the response did little to quell public scrutiny of the controversial approval. Critics were apt to point out that the FDA had a record of approving products in this manner. Years prior, the FDA had approved the drug Eteplirsen for the treatment of duchenne muscular dystrophy (DMD), despite the objections of their scientific staff which disclosed a lack of evidence for the efficacy of this treatment. Criticism was also directed at FDA leadership suggesting the approval of some medical products were potentially due to favoritism toward companies seeking approval.
Resolving Internal FDA Disagreements
While it is acknowledged that the FDA must consider technological and political implications alongside scientific evidence in decision-making, it is essential to address the concerns of these organizations and coalitions. As part of an ongoing project to reform the FDA’s Advisory Committee system and assist the FDA with getting the advice needed to make the best evidence-based decisions, FAS engaged in discussions with current and former Advisory Committee members to seek their input on resolving disagreements between FDA staff and leadership. These conversations highlighted the breach of trust between the FDA, its staff, and the public.
“FDA leadership needs to make is clear what data was used and why they’re moving forward when there is opposition”
“Disagreements should be addressed by a non-biased source because it affects the public safety.”
“There will be times where there are disagreements between staff and leadership. However, there’s a critical need for transparency within the FDA about why decisions are made. These are not decisions about evidence only. Ever.”
“Disagreements should be a matter of public scrutiny. There should be transparency that doesn’t jeopardize confidentiality.”
The FDA’s Commissioner has acknowledged the lack of trust within the institution and expressed a commitment to address the issue. There are many reasons for the lack of trust. However, one reason stems from the FDA’s approval of medical products despite clear opposition from its scientists and sometimes Advisory Committee members.
This raises the question of how the FDA intends to address these internal disagreements, which have the potential to impact the health and safety of the American public. Currently, the FDA has implemented a program called Scientific Dispute Resolution (SDR) to handle such conflicts. This document was initially developed in 2009 and recently updated in 2021 with the purpose of outlining the process for communication regarding internal scientific disputes within FDA Centers. It defines scientific disputes as disagreements that arise from the interpretation of science and the resulting decisions. This definition clearly distinguishes the circumstances in which the guidance should be utilized to resolve discrepancies between FDA scientists, statisticians, and their respective Center leadership. The guidance offers valuable examples of best practices for resolving formal and informal scientific disputes within the agency. Some of those best practices include, but are not limited to:
- Centers employing various mechanisms to disseminate dispute resolution Standard Operating Procedures (SOPs) to employees.
- Requiring Center SOPs for dispute resolution to clearly state that written documentation is the only avenue in which a formal dispute can be triggered (this step ensures appropriate historical documentation).
- Developing several avenues to address scientific issues through the use of regulatory briefings, advisory committees, internal discussions with Center Directors, standing subject matter committees, and multi-disciplinary teams.
However, it should be noted these best practice recommendations are not obligatory and their adoption is left to the discretion of the individual Centers. Furthermore, the document provides a process by which any of the internal parties involved can appeal the resolution of their dispute if they find it unsatisfactory through the Office of Research Integrity at Health & Human Services (HHS).
Policy Recommendations
To increase morale and improve the approach and resolution of internal disagreements within the agency, the policy recommendations are as follows:
- Ensure that all FDA staff and leadership are fully cognizant of the existence and details of the Scientific Dispute Resolution at FDA guidance and the process for submitting disputes for review.
- Incorporate the Scientific Dispute Resolution at FDA guidance into FDA regulations
- Note: To incorporate this guidance into FDA regulations, the FDA will propose the regulation for OMB review. OMB will review and open the regulation up for public comment through the Federal Register. Responses to comments will be developed, and a final draft submitted to OMB for review. If approved, the final regulation will be published in the Code of Federal Regulations (Congressional Research Service, 2013). The involvement of Congress will not be necessary.
- Amend the Scientific Dispute Resolution at FDA guidance to dictate the mandatory execution of best practices within the dispute resolution process.
- This guidance should identify additional non-biased parties (that may not be government-affiliated) to provide impartial guidance on complex scientific matters affecting public safety.
- Develop guidance that clearly explains a transparent process to communicate effectively with AdComm members regarding decision making when parties have opposing viewpoints
- Implementing a transparent process to communicate with AdComm members regarding differences between the agency and the AdComm will assist in improving morale between both parties, but also encourage continued support of the AdComm.
- This recommendation is also supported by a survey conducted by 3D Communications with 400+ AdComm members where 94% of members concurred that the FDA should develop a process to communicate their reasoning for decisions in opposition to Committee recommendations (3D Communications, 2024).
Conclusion
While FDA leadership ultimately holds the authority to grant approvals, it is crucial that the perspectives of all experts are duly considered. This includes the valuable input from the agency’s scientists, statisticians, and advisory committees. To regain public trust and restore integrity, it is imperative to first rebuild trust internally among the dedicated public servants within the FDA. Adoption of the aforementioned recommendations would start the trust-rebuilding process and lead to increased safety and precaution measures when approving drugs and medical devices.
Leveraging AdComm Membership
From January 2024 to July 2024, the Federation of American Scientists interviewed 30 current and former Advisory Committee (AdComm) members. Based on these discussions, we were able to source potential policy recommendations that may assist with enhancing the FDA’s ability to obtain valuable advice for evidence-based decision-making. The results of these discussions are presented in case study format detailing the recurring themes that emerged and policy recommendations for improvement.
The FDA relies on its scientific staff and Advisory Committees to provide conclusions from trial and study data, which aid in the process of regulating and approving medical products. Discussions have been centered around how to appropriately leverage the membership of Advisory Committee experts to assist with areas of difficulty surrounding the safety and efficacy of medical products. Nonetheless, the methods by which these systems currently generate the evidence the Government needs can be improved. This case study focuses on five key areas we believe can assist in fully utilizing the capacity in which AdComms serve and improve overall engagement with AdComms membership.
AdComm Membership Problems
Advisory Committees serve as the core for expert engagement in the Food and Drug Administration’s (FDA) decision-making processes and are composed of medical professionals, industry representatives, patient advocates, and scientific experts. Their primary role is to provide the FDA with informed advice and recommendations on issues spanning science, regulatory policy, and the evaluation of products under the FDA’s jurisdiction.
The intricacies of being an effective AdComm member, however, have been somewhat overlooked. Conversations with current and ex-members have highlighted areas for enhancement that would strengthen the function of AdComms and enrich the advice provided. Feedback indicated a lack of transparency in the FDA’s recruitment methods for committee positions, insufficient orientation or training for new members, limited understanding of regulatory procedures among members, and an onerous conflict of interest protocol that served as a deterrent for some members who were asked to return or renew their membership.
Pathways to Improving AdComms Membership
Committee Composition
The composition of Advisory Committees vary depending on the charter that has been set in place. In some cases, committee composition has been set by law. However, where there is flexibility in determining the composition of a committee, consideration should be given to all categories of expertise that should be included and diversity of voices that are selected to participate in these meetings. Committee composition should reflect the diversity of the world and populations of whom their recommendations could potentially affect. For this reason, discussions with current and former Advisory Committee members indicated the need for three additional areas of expertise that should be included on all Committees. Insights discovered that all Committees should include a patient representative who has the knowledge from lived experience and understanding of how treatments affect day-to-day life. This recommendation was further corroborated by a 3D Communications survey conducted with 400+ FDA AdComm members where results indicated that 48% agreed there should be a patient and consumer representative on all Committees. Members also stated that pharmacists should be included because drugs and devices eventually pass through their hands to give to patients. Pharmacologists should serve on the Committee due to their clinical application knowledge of drugs and devices. Finally, a roster of temporary members should be created for varying categories to use when additional expertise is needed on a Committee because of a conflict of interest or when a certain skillset or knowledge base is lacking on the current Committee.
Role of the Advisory Committee Chair
The FDA describes the purpose of an Advisory Committee chair as one who will “preside at committee meetings and ensure that all rules of order and conduct are maintained during each session”. The chair also has the responsibility of ensuring all recommendations and advice from AdComm members are clear and evidence-based. Moreover, the role of the chair should be used to enhance the overall committee experience as well as be of service to the FDA. Despite these requirements, there’s an underutilization of the chair’s role in terms of communication and stakeholder coordination, as evidenced by the chair not being listed as a primary point of contact for the Advisory Committee and a lack of coordination amongst stakeholders.
Chairpersons are usually selected due to their critical domain knowledge, understanding of best practices, ability to identify risks and keep members engaged, and expansive relationships within their industry. Maximizing the chairperson’s role requires discussion on how to utilize their valuable domain knowledge and professional networks. Chairs possess extensive networks that could support the identification of permanent or temporary expert participants for AdComms, aiding the FDA’s mission to recruit top talent for guidance. This would ensure the FDA’s continued success in recruiting the brightest minds in the industry to assist with providing advice. Additionally, chairs should have oversight in identifying relevant issues or products for their respective committees to appraise, which can provide another layer for the FDA to keep abreast of critical public concerns via appropriate committee evaluation.
Training
Training is a significant part of many Federal Government service positions. However, besides ethics and conflict of interest trainings, there is no set training program in place for most new Advisory Committee members. Considering Advisory Committee members come from different professional backgrounds with varying levels of expertise, the FDA should develop an onboarding training program to assist with acclimating all new AdComm members into their roles. Many former and current AdComm members mentioned that no formal training was provided as part of the onboarding process. Some members who were new to the FDA AdComm process or who were not physicians or scientists stated they had no knowledge of statistical analyses, clinical trial design, or how the FDA views the role of the AdComm in the regulatory process.
A foundational training, covering these aspects, would greatly benefit those members such as consumer and patient representatives who may lack this shared base of expertise. An investment in such an onboarding experience would promote stronger rapport among members and guarantee their preparedness in analyzing scientific and technical submissions.
Learning about the FDA Regulatory Process
The Food & Drug Administration (FDA) was established with the purpose of regulating drugs and medical devices to ensure their safety and effectiveness for all citizens in the United States. Many Advisory Committee members join these committees without basic knowledge of the FDA’s regulatory process. During FAS’ discussions with current and former AdComm members, approximately 71% of members stated that basic knowledge of the regulatory process and how the FDA makes their decisions was unknown to them.
Providing AdComm members with an introductory course on the FDA’s regulatory process could enhance their comprehension, potentially allowing them to make more effective contributions and informed clinical decisions (based on their occupation). Although the FDA provides some online resources about its processes, like FAQs and guidelines, an expansion of this material should be considered for inclusion in AdComm orientation activities.
Conflict of Interest (COI) Process
18 U.S.C. 208(a) prohibits Advisory Committee members who are designated as special government employees (SGE) from serving on federal advisory committees or any other Federal Government form of service that will have a “direct or predictable effect” on their financial interests. Similarly, the FDA describes a conflict of interest as an occurrence “when an individual selected to serve on an advisory committee has financial interests that may be impacted by the individual’s work on the advisory committee”. The auditing process for conflicts of interest is designed to confirm that the members of the advisory committee maintain impartiality and ensure the integrity of public health safety. Prior to any committee gathering, the FDA mandates that each participant, classified as either an AdComm member or SGE, complete an FDA 3410 form that reveals all financial connections that could be seen as potential COIs.
However, the process of what happens after the 3410 form has been completed is ambiguous. In 2007, the FDA submitted draft guidance to the federal register for comment entitled, Guidance for the Public, FDA Advisory Committee Members, and FDA Staff on Procedures for Determining Conflict of Interest and Eligibility for Participation in FDA Advisory Committees in an effort to determine if there is an inappropriate COI that should exclude members from participating in a committee meeting. Moreover, the official guidance is not easily accessible. Another draft guidance was developed with a detailed listing of considerations to be given when examining conflict of interests can be found in the FDA’s draft guidance on Procedures for Evaluating Appearance Issues and Granting Authorizations for Participation in FDA Advisory Committees.
Discussions with current and former AdComm members about the COI auditing process sparked varying views regarding whether flexibility should be exercised for COIs. 82% of members concurred that while they recognize the necessity for such a system, it tends to be overly demanding due to repetitive paperwork, especially when their circumstances remain unchanged. The strenuous nature of this routine has even deterred some from continuing their membership each year and remains a key aspect as to why members choose to end their service.
Despite having a COI process in place, there are loopholes that allow members with conflicts of interest to remain as voting members for specific AdComm meetings. A certain incident involved an Advisory Committee where 10 members who had financial ties to the sponsor were allowed to participate in an AdComm meeting. These individuals ultimately took part in endorsing the TriClip G4 System by Abbott and unanimously agreed that its benefits outweighed the potential risks. To further complicate matters, this information was not disclosed to the public at the time of approval.
While the COI process has resulted in members being rightfully disqualified from meetings due to actual or apparent conflicts, there is room to refine how these conflicts are identified and the standards employed to judge permissible COIs.
Policy Recommendations
In an effort to better leverage Advisory Committee membership, the potential policy recommendations are as follows:
Committee Composition
- If there is flexibility and committee composition is not bound by law, include a patient representative and pharmacist and/or pharmacologist on each Committee
- Patient representatives provide a needed perspective due to lived experiences and understanding of how specific drugs and devices affect their day-to-day life
- Drugs and devices will usually pass through the hands of pharmacists and pharmacologists. Therefore, they should have the opportunity to serve on these Committees and provide feedback. Pharmacologists also understand the clinical application of drugs.
- Develop a register of temporary members that can be utilized when additional expertise is needed for Committee meetings or when there is a conflict of interest (COI)
Role of the Chair
- Expand the role of the Committee chair that will encompass the task of recruiting both standing and ad hoc members, as well as identifying prominent issues and products for Committee consideration, thereby allowing for specialized input from their Committee
Training and Regulatory Process
- Institute a basic 101 training for all newly appointed Advisory Committee members that covers statistical analysis, clinical trial design, and elucidates the partnership between the FDA and AdComm
- Include an overview of the regulatory process and how the FDA’s decision-making process is performed
COI Auditing Process
- Develop a process that can quickly replace individuals who have a known conflict of interest
- Clearly delineate criteria for committee service acceptance regarding individuals with potential or actual conflicts of interest
- Streamline the COI process to prevent duplicative work that may act as a deterrent to experts volunteering to serve on the AdComm (work with GSA on this matter if necessary)
Conclusion
Advisory Committees are pivotal to maintaining trust with the public. It is essential for public safety to ensure that the most qualified experts are selected to serve on these Committees and that they have the tools to provide the FDA with informed and evidence-based recommendations. In an effort to increase public health safety, the FDA should enhance the AdComm structure by expanding the chair’s role, creating training programs for all new Advisory Committee members, and revising the conflict of interest procedures.
The Future of Voting for FDA Advisory Committees
From January 2024 to July 2024, the Federation of American Scientists interviewed 30 current and former Advisory Committee (AdComm) members. Based on these discussions, we were able to source potential policy recommendations that may assist with enhancing the FDA’s ability to obtain valuable advice for evidence-based decision-making. The results of these discussions are presented in case study format detailing the recurring themes that emerged and policy recommendations for improvement.
Advisory Committees (AdComms) serve as the core for expert engagement in the Food and Drug Administration’s (FDA) decision-making processes. These committees are composed of medical professionals, industry representatives, patient advocates, and scientific experts. Their primary role is to provide the FDA with informed advice and recommendations on issues spanning science, regulatory policy, and the evaluation of products under the FDA’s jurisdiction. Public meetings led by the FDA with these committees are instrumental in facilitating transparent deliberation between the FDA, the advisory body, and the American public. This practice helps to cultivate a collaborative environment between the FDA, the AdComms, and the public. AdComm recommendations are integral to strengthening public trust and reinforcing the FDA’s credibility. This relationship is corroborated by aligning the counsel of these independent entities with the FDA’s regulatory actions.
Key Problems Facing Advisory Committees
A critical component of the AdComm structure is its voting mechanism, a method by which hand-selected experts offer expert advice or recommendations on questions that have been proposed by the FDA to assist with informing its formal, regulatory decision-making. These questions include a broad range of topics, from evaluating post-market safety data to assessing pre-market product risks and benefits, and gauging whether a product should be approved or withdrawn from the market. The outcomes of the votes serve as barometers for the AdComms official stance on products and provide the FDA with a comprehensive and collective viewpoint. However, the recommendations proposed by AdComms are suggestive rather than prescriptive; ultimately, leaving the final decision to FDA leadership.
Recent patterns indicate a reduction in the convening of AdComm meetings. In 2010, 55% of FDA-approved drugs were referred to an advisory committee. By 2021, the percentage of FDA-approved drugs with an advisory committee referral had dropped to 6%. The decline of meetings eliminates opportunities for evidence-based evaluation and deliberation that could potentially affect the health and well-being of Americans. Furthermore, the diminishing of these crucial interactions between the FDA, AdComms and the public exacerbates the lack of trust and erodes transparency. Interestingly, while most committees present definitive votes that are supported by explicit justifications for either approving or rejecting items under review, FDA Commissioner Robert Califf has suggested in multiple interviews that AC votes can be useful but are not mandatory for every meeting. This viewpoint raises concerns about the potential removal of voting from the reform agenda, which could undermine AdComms capacity to evaluate intricate topics that affect the American public consumer base. In addition, a survey conducted by 3D Communications with 400+ current and former AdComm members asked about the importance of voting. Results showed that 95% of AdComm members believe that voting should be retained when reviewing the benefits and risks of medical products. Reform discussions have materialized due to these factors, in addition to the FDA’s accelerated approval of Adulhelm (aducanumab) despite clear AdComm opposition. Demand for reform is inevitable and many are urging for there to be an increased number of AC meetings and a thorough reorganization of advisory committee operations and voting protocols. Such reform is not only administrative but also symbolic. This type of reform confirms that decisions affecting public health should be informed and shaped by multidisciplinary expertise. Additionally, it re-establishes the pivotal role of public input in regulatory affairs, which is an indispensable component to maintaining the American public’s trust.
Significance of AdComm Voting
In response to this call for AdComm reform, a project spearheaded by FAS has sought feedback from AdComm members regarding their views on the significance of voting. The intention of engagement is to understand members’ experiences as experts and their perspectives on voting by asking the following questions:
- Describe your Advisory Committee’s voting process.
- On a 1-5 scale, how crucial is it for you to vote on products?
These questions aimed to measure the variability in voting mechanisms across committees and the value members place on voting. Results from 30 participants demonstrate a consensus on the critical role of voting in formulating committee recommendations, with 87% of committee members indicating a five (very important) as their stance on the importance of voting.
Policy Recommendations
To uphold the FDA’s integrity and regain public confidence, retaining voting at AdComm meetings is essential in addition to other recommendations to enhance the advisory committee process. The recommendations are as follows:
- Maintain voting as an integral function, allowing FDA Advisory Committee members to convey their collective expertise and advice, aiding the FDA in informed decision-making on scientific and regulatory matters.
- Revise the guidance for FDA Advisory Committee Members and FDA Staff to explicitly define circumstances for which voting should occur, eliminate sequential voting
Conclusion
The recommendations of FDA Advisory Committee members are a pivotal component to the FDA’s regulatory decision-making process. Maintaining the voting protocol for Advisory Committee meetings is essential as members strive toward the continued provision of precise, impartial, and evidence-based counsel to the FDA. This voting mechanism guarantees the inclusion of each member’s perspective and ensures that an official committee stance is taken, offering the FDA definitive and straightforward guidance.