Scientists take action by engaging with Congress on the promise of monoclonal antibody therapies for COVID-19
The limited science and technology (S&T) resources available to policymakers in Congress and state legislatures have compounded the severity of the COVID-19 pandemic. To help meet legislators’ S&T needs, the Federation of American Scientists (FAS) organized more than 60 specialists with expertise in all the different aspects of the pandemic to serve on the COVID-19 Rapid Response Task Force. In addition to providing many written briefings to Congressional and state legislative offices, members of the Task Force have so far provided three oral briefings to Congress, including one about the promise of monoclonal antibody (mAb) therapies for COVID-19.
Monoclonal antibodies to counter COVID-19
Many experts believe that mAb therapies have a major role to play in protecting people from COVID-19, serving as a bridge to a vaccine, and safeguarding groups that potentially would not respond to a vaccine, for example, an aged population or immunocompromised individuals. There is certainly precedent for mAb therapies helping people survive deadly diseases; for instance, mAb therapies resulted in statistically significant survival benefits for Ebola patients.
Therapeutic mAbs against COVID-19 are intended to stick to the spike proteins of SARS-CoV-2, which are on the outside of the virus, and block them from being able to attach to receptor proteins on the outside of human cells, which would be needed for viral entry and infection. Most focus has been on the spike protein interaction with the human cell surface receptor ACE2, and recent work shows that the human cell surface protein Neuropilin-1 also has a role in facilitating SARS-CoV-2 entry and infectivity in some human cell types. Designing mAb therapies that interfere with either one or both of these interactions could help explore the range of efficacies of COVID-19 mAbs.
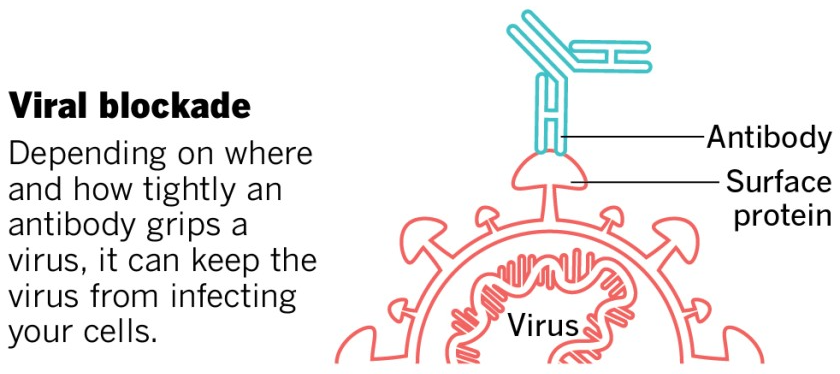
This image was adapted from the San Diego Union-Tribune.
An experimental mAb from Eli Lilly being tested in early-stage clinical trials, while not appearing to help COVID-19 patients who are hospitalized (that testing has been stopped), has shown promise for people with mild or moderate COVID-19 who receive the treatment early and who are not hospitalized, reducing viral load, symptom severity, and eventual hospitalizations. The Food and Drug Administration (FDA) recently granted an Emergency Use Authorization (EUA) for Eli Lilly’s antibody. Under the EUA, the treatment, called bamlanivimab, is supposed to be given to patients as soon as possible after a positive coronavirus test, no more than 10 days after developing symptoms. The treatment should not be used for patients who are hospitalized. It is intended for individuals 12 and older and at risk for developing a severe form of COVID-19 or being hospitalized.
Regeneron has also developed a mAb treatment, and has, like Eli Lilly, stopped its clinical trial in hospitalized patients – in Regeneron’s case, “an independent data monitoring committee warned that the risks might outweigh the benefits for hospitalised patients on high levels of oxygen.” Like Eli Lilly’s drug, Regeneron has reported that its mAB cocktail reduces virus levels in the body and improves symptoms for individuals with COVID-19 who are not hospitalized. Regeneron has also applied to the FDA for an EUA of their mAB therapy, and overall, at least ten COVID-19 antibodies are being tested in clinical trials, with many more under development.
While EUAs can serve to get drugs to patients more rapidly than going through full FDA approval, the use of mAbs outside of clinical trials can make it more difficult to ascertain the therapies’ true effectiveness in different age groups. Furthermore, it could make it harder to enroll volunteers in future clinical trials for alternative therapies, since people may want to take a drug that appears to work, rather than risk possibly getting placebo. Authorizing a low-impact therapeutic could be counterproductive. FDA must take care to only grant EUAs for mAb therapies where the data show they are potent, and clearly delineate the circumstances in which they should be administered to help patients.
Antibodies are expensive and difficult to make, and they are administered at relatively high levels. These factors conspire to limit the number of doses that are produced. By the end of the year, Regeneron is expected to have produced up to 300,000 doses of its cocktail, and Eli Lilly greater than one million. More doses are needed; experts estimate that each day, 10,000 to 15,000 people in the US would be indicated for the drugs based on age and risk factors, even if there’s no further surge of infection.
A major challenge is that manufacturing capacity for monoclonal antibodies is limited, generally actively being used to produce treatments that are needed by patients as therapies for conditions such as cancer, multiple sclerosis, or osteoporosis. Globally, bioreactor capacity for producing mAbs from mammalian cells is spread across 200 facilities, with a total capacity of about 1,500,000 gallons. Constructing new manufacturing capacity requires years, with different types of facilities taking anywhere from 18 months to 7.5 years to come online. Bringing inactive facilities back online is a possibility, but how available such facilities are, and what would be needed to update them for operation, is unclear, and unlikely to happen in the near future. So, COVID-19 mAb manufacturing will need to rely on facilities either in use or in development, at least in the near-term.
Possible roles for government include helping to coordinate the construction of new mAb manufacturing capacity, or the repurposing of existing sites that could serve as bioreactors. Non-traditional mammalian cell lines could be tested to see which produce the highest levels of these mAbs to make the best use of the bioreactor capacity. Also, there are techniques for producing mAb therapies in bacterial or fungal (like brewer’s yeast) cells, in addition to mammalian cells, which could take advantage of existing capacity for microbial fermentation. And regulatory agencies like the Environmental Protection Agency and FDA could help expedite actions like repurposing bioreactor sites or deploying new bioproduction technologies by prioritizing the evaluation of these activities without harming the environment or sacrificing drug safety or efficacy.
Government could also assist with coordination between companies, providing a framework and forum for sharing information about or partnering on manufacturing capacity, or discussing approaches to COVID-19 mAb design that may have already been attempted and that did not pan out. This coordination could even extend to facilitating international collaborations, as COVID-19 mAb development efforts are taking place in countries all around the globe.
Considering COVID-19 is a deadly disease that can also have long-term health impacts for those who survive, the development and deployment of mAb therapies that can be administered soon after infection is detected and reduce the severity of disease are expected to contribute to countering the health effects of the pandemic.
Briefers
The experts who briefed Congress were Megan Coffee, MD/PhD, Clinical Assistant Professor at New York University; Erica Ollmann Saphire, PhD, Professor at the La Jolla Institute for Immunology and lead of the Coronavirus Immunotherapy Consortium; Jill Horowitz, PhD, Executive Director of the Strategic Operations Laboratory of Molecular Immunology at Rockefeller University; John Cumbers, PhD, CEO of SynBioBeta; Eric Hobbs, PhD, CEO of Berkeley Lights; Jake Glanville, PhD, CEO of Distributed Bio; Mike Fisher, PhD, Senior Fellow at the Federation of American Scientists; and Ali Nouri, PhD, President of the Federation of American Scientists.For more information about FAS’ work with Congress, visit our Congressional Science Policy Initiative website.
Categories: Public Health
The NCARS Act would amend the National Security Act of 1947 to establish a durable, coordinated federal approach to national resilience.
Nearly 150 organizations and government officials have endorsed the call to action and solutions for extreme heat, now public at HeatAgenda.US Washington, D.C. – July 7, 2026 – As millions of Americans continue to struggle to stay cool following one of the hottest Independence Day holidays on record, the Federation of American Scientists (FAS), one […]
Addressing rising heat will take all of us. Together, we can create heat-safe homes, workplaces, schools, childcare facilities, and communities – the backbone of a heat-ready nation.
If you’re new to the climate intervention space, welcome! The TL;DR: if we can’t stop the most catastrophic impacts of climate change with current tools quickly enough, then we need a bigger toolbox.