Your DNA, Your Data: Preventing Genetic Discrimination in the Growing Bioeconomy
The U.S. bioeconomy is a growing economic sector driving technological innovation and global competitiveness. A significant portion of this innovation, especially in biotechnologies that improve health, like drug therapeutics and precision medicines, relies on the collection of genetic and non-genetic biological information through varied methods, including academic studies, direct-to-consumer testing services, and pharmaceutical companies. While this can lead to improvements in U.S. public health and biotechnology, there has been a growing fear among scientists and legal experts that this information is insufficiently protected against exploitation by foreign actors seeking to supplant U.S. leadership in biotechnology, as well as against domestic actors who might use this data to target or discriminate against certain subsets of the population
Legislation and policy outlining the storage and use of human-derived biological data by federally-funded research, such as the NIH Genomic Data Sharing Policy, lessen the risks surrounding this data but are insufficient due to advancements in biotechnology and the multifaceted collection, use, and selling of this information by private industry and law enforcement. Meeting the moment and protecting the American people will require: 1) expanded legislative protections for biological data; 2) biological data use protocols developed for federal agencies; and 3) standardized development, storage, and use of biological data. Pursuing these policy enhancements will safeguard fundamental rights and secure national infrastructure as we enter a new era of biological understanding and innovation.
Challenge and Opportunity
The U.S. bioeconomy is an increasingly important facet of the GDP due to the growing role of biotechnology in economic sectors, including defense, agriculture, energy, and manufacturing, with the total market size of biotechnology expected to reach $2-4 trillion in 2030-40. An important driver of this growth is the increased role of biotechnology in drug discovery, therapeutics development, and precision medicine.
This innovation, which includes novel treatments for cystic fibrosis, has necessitated the collection of massive datasets of biological data, including genetic, molecular, and biometric information. The federal government supports this through the direct creation and management or funding through grants of large-scale biobanks of individuals from varied geographic, demographic, and health backgrounds to be primarily used in biomedical research. Various pharmaceutical, technology, and biotechnology companies have additionally collected millions of primarily genetic samples from members of the public; for example, more than 26 million people have taken direct-to-consumer genetic tests through companies, such as 23andMe and Ancestry.
However, as more human-derived biological datasets grow, they become strategic targets. There is an increasing concern that this information is insufficiently protected against exploitation by foreign actors seeking to supplant U.S. biotechnological leadership, as well as against malicious domestic actors who may misuse biological data to perpetrate genetic discrimination and biological data discrimination. While this may initially seem like a concern limited to employment or insurance, genetic discrimination has potential negative ramifications for the unchecked surveillance and intrusion into the private lives of Americans. Cases of genetic discrimination have already been identified in education, such as the case of Colman Chadam, a middle-schooler forced to leave his school because of a genetic susceptibility to cystic fibrosis. Additional civil liberties concerns arise around the non-consensual misuse of biological data in law enforcement investigations. Even while measures have been taken to secure biological datasets to minimize the number of people who might misuse this information, both public and private data collections are under scrutiny from sectors of the public about the risk of anonymized biological data being reidentified and the ability of these collectors to prevent data leaks.
The preeminent federal law guiding the use of this data in non-research settings is the Genetic Information Nondiscrimination Act (GINA). GINA, in combination with the Health Insurance Portability and Accountability Act (HIPAA) and other legislation, outlines the general use of genetic and biological information within the U.S. However, these laws leave regulatory gaps that enable the previously mentioned civil rights violations to rise. HIPAA only provides protections to biological information in the context of “protected health information” for covered entities, such as healthcare providers and their business associates. And GINA only prohibits the use of genetic data to discriminate in employment and health insurance coverage, and only restricts organizations with greater than 15 employees. Moreover, there are no laws that protect against discrimination surrounding the use of non-genetic biological information, such as those collected by private companies and personal health trackers. Protections beyond GINA at the state level are inconsistent and lacking, especially given the highly personal and largely unchanging nature of this information. While guidance exists for the storage, sharing, and oversight of biological data for institutions that receive federal funding, there is a lack of this same technical standard for commercial and direct-to-consumer companies.
This regulatory vacuum allows deeply personal information about an individual’s disease history, familial relationships, and potential traits to be used to cause harm. This opens the door to dangerous infringements on personal safety and human rights, threatening the stability of the growing bioeconomy.
Plan of Action
To secure the U.S. bio-infrastructure, maintain global leadership in biotechnology, and safeguard American citizens from emerging threats to their privacy, the federal government must modernize its approach to human genetic and biological data. The current regulatory patchwork leaves the bioeconomy vulnerable to foreign exploitation and American citizens open to unchecked surveillance. The following recommendations establish a necessary framework to build trust in U.S. innovation while protecting individual liberty.
Recommendation 1. Modernize Genetic Privacy Laws to Close Security Gaps
Congress should advance legislation that comprehensively expands GINA to include all forms of biological data, including but not limited to: genetic, protein, microbiome, and biometric data, in order to close the loopholes present in the original law.
To ensure that an American’s biological information remains their private property and not a tool of overreach, this legislation should expand nondiscrimination protections beyond health insurance and employment. This legislation could be modeled on CalGINA, a 2011 California law that adds “genetic information” to existing protected classes, such as race, sex, and age. New federal legislation would expand on this model by codifying “biological information” as a protected class with “genetic information” within existing civil rights law. Additionally, this legislation should include direct language from CalGINA that prevents business establishments, health facilities, housing providers, and state-funded programs from demanding genetic tests, and penalizing Americans for their biological makeup. This legislation can also use language from the EU’s General Data Protection Regulation (GDPR) that classifies genetic and biometric data under a special “sensitive data” category. GDPR language would assist in classifying the scope of genetic and biological data as well as the protections individuals possess in regards to this information.
By setting this new federal baseline, Congress will harmonize the current fragmented regulatory landscape, clarifying compliance for businesses that may seek this information, and will assure the American public that their biological information cannot be weaponized against them.
Recommendation 2. Establish Guidelines For The Federal Use Of Biological Data
To prevent unwarranted surveillance and privacy erosion, the president should issue a memorandum tasking the National Institute of Standards and Technology (NIST) and the Office of Science and Technology Policy (OSTP) with developing a “Federal Human-Derived Biological Data Use Standard”.
To ensure the standard accounts for the full spectrum of federal use cases, NIST and OSTP, in coordination with the Office of Management and Budget (OMB) and the National Security Council, must conduct an interagency review of all current and potential federal uses of biological data. The Standard should specifically adopt a privacy-centric model, similar to that established by the 2019 Interim Department of Justice policy on genetic genealogy. Once developed, federal agencies must make the Standard available to state and local partners to serve as a model for non-federal policy. Additionally, OSTP should publish a public-facing framework that clarifies federal use cases. This framework must include clear definitions of biological data types, transparent access standards, a list of actions explicitly prohibited by the new protocol, and clear accountability mechanisms.
This standard will define strict policies for permissible federal use of biological data to streamline disparate protocols and prevent the over-exposure of citizen data by the federal government. It will additionally serve as a model to ensure consistent protection for Americans across all levels of government.
Recommendation 3. Implement Technical Standards For Biological Data Security And Innovation
The president should direct OMB to issue a Biological Data Protection Directive. This directive must mandate that federal agencies standardize the technical infrastructure regarding how human-derived biological data is collected, stored, and shared.
Specifically, the Directive should:
- Develop National Security Standards. Task the National Science and Technology Council (NSTC) to form a subcommittee that works with NIST to develop national technical standards for data storage, sharing, and selling. NIST should leverage recommendations from the National Security Commission on Emerging Biotechnology to ensure these standards promote AI-readiness and interoperability.
- Leverage Federal Buying Power. Direct the National Institutes of Health (NIH) to condition federal grant funding on compliance with these updated NIST data security and interoperability standards.
- Support Private Sector Adoption. Direct the Federal Trade Commission (FTC) to engage industry stakeholders to identify technical obstacles preventing the private sector from adopting NIST standards. The FTC should collaborate with stakeholders to build technical roadmaps for adoption and create a public database of companies that fail to update their protocols or adhere to these security standards.
To implement these activities, the president should request Congress to appropriate $50-80 million over three years for staffing, training, and technical infrastructure. Standardizing this infrastructure will close security gaps that currently allow foreign adversaries to target American biological data while driving market-wide adoption of secure protocols and reducing friction for U.S. businesses.
Conclusion
Biological data is becoming as central to modern society as a traditional digital footprint and carries similar far-reaching risks if misused. Without proactive federal action, the expanding role of biological data will continue to enable new forms of discrimination, privacy violations, and civil-rights harms, while leaving critical national assets vulnerable to exploitation by foreign competitors and unchecked domestic surveillance.
If successfully and fully implemented, these new policies would protect individual rights and secure the bioeconomy, establishing the United States as a leader in responsible biotechnology innovation. The first recommendation would provide clear and enforceable civil protections for all Americans, ensuring that individuals, businesses, and institutions cannot misuse biological information regardless of how it was obtained. This would prevent cases like that of Colman Chadam from recurring. The second recommendation would support more effective and accountable law enforcement by establishing rigorous, updated guidelines that limit federal overreach, and ultimately reduce privacy risks while improving public trust. Finally, the third recommendation would strengthen federally funded and private biomedical research by developing standards that make biological data interoperable, AI-ready, and secure.
The combination of all the recommendations will provide clarity to both state and private actors on appropriate development, storage, and use of biological information. This approach ensures that U.S. values define the global bioeconomy, creating lasting protections for the use of this information in critical facets of society.
The proposed legislation will define “biological information” broadly to encompass all data derived from biological samples or measurements that can reveal health, behavioral, or ancestry-related traits. This includes molecular, biometric, and physiological data that can infer or predict personal or familial traits and diseases.
The proposal complements, rather than replaces, existing civil rights frameworks. By adding “biological information” to the list of protected classes, the law provides a clear and enforceable basis for addressing discrimination that current statutes do not explicitly cover.
Enforcement will rely on existing federal civil rights and consumer protection infrastructure. The Equal Employment Opportunity Commission will be empowered to investigate biological-data–based discrimination in employment, while the Department of Justice’s Civil Rights Division can address systemic violations across public institutions. The Federal Trade Commission will continue to regulate unfair or deceptive data practices by private companies.
Many small businesses and startups are already taking scattered approaches to protecting their data. This policy would remove burdens and accelerate biotechnological innovation by providing clear standards for the use of biological data for those entering the field and lowering the delays necessary to understand a scattered regulatory landscape.
Due to advancements in biotechnology, malicious domestic actors may use biological data to target, blackmail, or exploit different segments of the American public who have voluntarily provided this information. This policy would minimize those risks by securing personal information and provide clear ramifications for misuses.
By implementing this policy, the U.S. will be the first country to make comprehensive policy on the security and use of personal biological datasets by federal and private actors. This policy will thus serve as a model for other nations realizing the dangers and necessity of protecting this type of information.
Making Healthcare AI Human-Centered through the Requirement of Clinician Input
Through partnership with the Doris Duke Foundation, FAS is advancing a vision for healthcare innovation that centers safety, equity, and effectiveness in artificial intelligence. Informed by the NYU Langone Health symposium on transforming health systems into learning health systems, FAS seeks to ensure that AI tools are developed, deployed, and evaluated in ways that reflect real-world clinical practice. FAS is leveraging its role in policy entrepreneurship to promote responsible innovation by engaging with key actors in government, research, and software development. These recommendations align with emerging efforts across health systems to integrate human-centered AI and evidence-based decision-making into digital transformation. By shaping AI grant requirements and post-market evaluation standards, these ideas aim to accelerate safe, equitable implementation while supporting ongoing learning and improvement.
The United States must ensure AI improves healthcare while safeguarding patient safety and clinical expertise. There are three priority needs:
- Embedding clinician involvement in the development and testing of AI tools
- Using representative data and promoting human-centered design
- Maintaining continuous oversight through post-market evaluation and outcomes-based contracting
This memo examines the challenges and opportunities related to integrating AI tools into healthcare. It emphasizes how human-centered design must ensure these technologies are tailored to real-world clinical environments. As AI adoption grows in healthcare, it is essential that clinician feedback is embedded into the federal grant requirements for AI development to ensure these systems are effective and aligned with real-world needs. Embedding clinician feedback into grant requirements for healthcare AI development and ensuring the use of representative data will assist with promoting safety, accuracy, and equity in healthcare tools. In addition, regular updates to these tools based on evolving clinical practices and patient populations must be part of the development lifecycle to maintain long-term reliability. Continuous post-market surveillance is necessary to ensure these tools remain both accurate and equitable. By taking these steps, healthcare systems can harness the full potential of AI while safeguarding patient safety and clinician expertise. Federal agencies such as the Office of the National Coordinator for Health Information Technology (ONC), the Food and Drug Administration (FDA) can incentivize clinician involvement through outcomes-based contracting approaches that link funding to measurable improvements in patient care. This strategy ensures that grant recipients embed clinician expertise at key stages of development and testing, ultimately aligning incentives with real-world health outcomes.
Challenge and Opportunity
The use of AI tools such as predictive triage classifiers and large language models (LLMs) have the potential to improve care delivery. However, there are significant challenges in integrating these tools effectively into daily clinical workflows without meaningful clinician involvement. As just one example, AI tools used in chronic illness triage can be particularly useful in helping to prioritize patients based on the severity of their condition, which can lead to timely care delivery. However, without direct involvement from clinicians in validating, interpreting, and guiding AI recommendations, these tools can suffer from poor usability and limited real-world effectiveness. Even highly accurate tools can become irrelevant if they are not adopted and clinicians do not engage with them, thereby reducing the positive impact they can have on patient outcomes.
Mysterious Inner Workings
The mysterious box of AI has fueled skepticism among healthcare providers and undermined trust among patients. Moreover, when AI systems lack clear and interpretable explanations, clinicians are more likely to avoid or distrust them. This response is attributed to what’s known as algorithm aversion. Algorithm aversion occurs when clinicians lose trust in a tool after seeing it make errors, making future use less likely, even if the tool is usually accurate. Designing AI with human-centered principles, particularly offering clinicians a role where they can validate, interpret, and guide AI recommendations, will help build trust and ensure decisions remain grounded in clinical expertise. A key approach to increasing trust and usability would be institutionalizing clinician engagement in the early stages of the development process. By involving clinicians during the development and testing phases, AI developers can ensure the tools fit seamlessly into clinical workflows. This will also help to mitigate concerns about the tool’s real-world effectiveness, as clinicians will be more likely to adopt tools they feel confident in. Without this collaborative approach, AI tools risk being sidelined or misused, preventing health systems from becoming genuinely adaptive and learning oriented.
Lack of Interoperability
A significant challenge in deploying AI tools across healthcare systems is the issue of interoperability. Most patients receive care across multiple providers and healthcare settings, making it essential for AI tools to be able to seamlessly integrate with electronic health records (EHR) and other clinical systems. Not having this integration could lead to tools losing their clinical relevance, effectiveness, and ability to be adopted on a larger scale. This lack of connectivity can lead to inefficiencies, duplicate testing, and other harmful errors. One way to address this is through a contracting process called Outcomes-based contracting (OBC), discussed shortly.
Trust in AI and Skill Erosion
Beyond trust and usability, there are broader risks associated with sidelining clinicians during AI integration. The use of AI tools without clinician input also presents the risk of clinician deskilling. Deskilling refers to the occurrence where clinicians’ skills and decision-making abilities erode over time due to their reliance on AI tools. This skill erosion leads to a decline in the judgement in situations where AI may not be readily available or suitable. Recent evidence from the ACCEPT trial shows that endoscopists’ performance dropped in non-AI settings after months of AI-assisted procedures. This presents a troubling phenomenon that we should aimt to prevent. AI-induced skill erosion also raises ethical concerns, particularly in complex environments where over-reliance on AI could erode clinical judgement and autonomy. If clinicians become too dependent on automated outputs, their ability to make critical decisions may be compromised, potentially impacting patient safety.
Embedded Biases
In addition to the erosion of human skills, AI systems also risk embedding biases if trained on unrepresentative data, leading to unfair or inaccurate outcomes across different patient groups. AI tools may present errors that appear plausible, such as generating nonexistent terms, which pose serious safety concerns, especially when clinicians don’t catch those mistakes. A systematic review of AI tools found that 22% of studies involved clinicians throughout the development phase. This lack of early clinician involvement has contributed to usability and integration issues across AI healthcare tools.
All of these issues underscore how critical clinician involvement is in the development of AI tools to ensure they are usable, effective, and safe. Clinician involvement should include defining relevant clinical tasks, evaluating interpretability of the system, validating performance across diverse patient groups, and setting standards for handoff between AI and clinician decision-making. Therefore, funding agencies should require AI developers to incorporate representative data and meaningful clinician involvement in order to mitigate these risks. Recognizing these challenges, it’s crucial to understand that implementing and maintaining AI requires continual human oversight and substantial infrastructure. Many health systems find this infrastructure too resource-intensive to properly sustain. Given the complexity of these challenges, without adequate governance, transparency, clinician training, and ethical safeguards, AI may hinder rather than help the transition to an enhanced learning health system.
Outcome-based Models (OBM)
To ensure that AI tools deliver properly, the federal contracting process should reinforce clinical involvement through measurable incentives. Outcomes-based contracting (OBC), a model where payments or grants are tied to demonstrated improvements in patient outcomes, can be a powerful tool. This model is not only a financing mechanism, but serves as a lever to institutionalize clinician engagement. By tying funding to real-world clinical impact, this compels developers to design tools that clinicians will use and find value in, ultimately increasing usability, trust, and adoption. This model provides a clear reward for impact rather than just for building tools or producing novel methods.
Leveraging outcomes-based models could also help institutionalize clinician engagement in the funding lifecycle. This would ensure developers demonstrate explicit plans for clinician participation through staff integration or formal consultation as a prerequisite for funding. Although AI tools may be safe and effective at the initial onset of their use, performance can change over time due to various patient populations, changes in clinical practice, and updates to software. This is known as model degradation. Therefore, a crucial component of using these AI tools needs to be regular surveillance to ensure the tools remain accurate, responsive to real-world use with clinicians and patients, and equitable. However, while clinician involvement is essential, it is important to acknowledge that including clinicians in all stages of the AI tool development, testing, deployment, and evaluation may not be realistic given the significant time cost for clinicians, their competing clinical responsibilities, and their limited familiarity with AI technology. Despite these factors, there are ways to engage clinicians effectively at key decision points during the AI development and testing process without requiring their presence at every stage.
Urgency and Federal Momentum
Major challenges associated with integrating AI into clinical workflows due to poor usability, algorithm aversion, clinician skepticism, and the potential for embedded biases in these tools highlight a need for thoughtful deployment of these tools. These challenges have presented a sense of urgency in light of recent healthcare shifts, particularly with the rapid acceleration of AI adoption after the COVID-19 pandemic. This drove breakthroughs in the areas of telemedicine, diagnostics, and pharmaceutical innovation that simply weren’t possible before. However, with the rapid pace of integration also comes the risk of unregulated deployment, potentially embedding safety vulnerabilities. Federal momentum supports this growth, with directives placing emphasis on AI safety, transparency, and responsible deployment, including the authorization of over 1,200 AI powered medical devices, primarily used in radiology, cardiology, and pathology, which tend to be areas that are complex in nature. However, without clinician involvement and the use of representative data for training, algorithms for devices such as the ones mentioned may remain biased and fail to integrate smoothly into care delivery. This disconnect could delay adoption, reduce clinical impact, and increase the risk of patient harm. Therefore, it’s imperative we set standards, embed clinician expertise in AI design, and ensure safe, effective deployment for the specific use of care delivery.
Furthermore, this moment of federal momentum aligns with broader policy shifts. As highlighted by a recent CMS announcement, the White House and national health agencies are working with technology leaders to create a patient-centric healthcare ecosystem. This includes a push for interoperability, clinical collaboration, and outcomes-driven innovation, all of which bolster the case for clinician engagement being woven into the very fabric of AI development. AI can potentially improve patient outcomes dramatically, as well as increase cost-efficiency in healthcare. Yet, without structured safeguards, these tools may deepen existing health inequities. However, with proper input from clinicians, these tools can reduce diagnostic errors, improve accuracy in high-stakes cases such as cancer detection, and streamline workflows, ultimately saving lives and reducing unnecessary costs.
As AI systems become further embedded into clinical practice, they will help to shape standards of care, influencing clinical guidelines and decision-making pathways. Furthermore, interoperability is essential when using these tools because most patients receive care from multiple providers across systems. Therefore, AI tools must be designed to communicate and integrate data from various sources, including electronic health records (EHR), lab databases, imaging systems, and more. Enabling shared access can enhance the coordination of care and reduce redundant testing or conflicting diagnoses. To ensure this functionality, clinicians must help design AI tools that account for real-world care delivery across what is currently a fragmented system.
Reshaping Healthcare AI
These challenges and risks culminate in a moment of opportunity where we can reshape and revolutionize the way AI supports healthcare delivery to ensure that its design is trustworthy and focused on outcomes. To fully realize this opportunity, clinicians must be embedded into various stages of AI development technology to improve its safety, usability, and adoption in healthcare settings. While some developers do involve clinicians during development, this practice is not the standard. Bridging this gap requires targeted action to ensure clinical expertise is consistently incorporated from the start. One way to achieve this is through federal agencies requiring AI developers to integrate representative data and clinician feedback into their AI tools as a condition of funding eligibility. This approach would improve the usability of the tool and enhance its contextual relevance to diverse patient populations and practice environments. Further, it would address current shortcomings as evidence has shown that some AI tools are poorly integrated into clinical workflows, which not only reduces their impact, but also undermines broader adoption and clinician confidence in the systems. Moreover, creating a clinician feedback loop for these systems will reduce the clerical burden that many clinicians experience and allow them to spend more dedicated time with their patients. Through the incorporation of human-centered design, we can mitigate issues that would normally arise by using clinician expertise during the development and testing process. This approach would build trust amongst clinicians and improve patient safety, which is most important when aiming to reduce errors and misinterpretations of diagnoses. With strong requirements and funding standards in place as safeguards, AI can transform health systems into adaptable learning environments that produce evidence and deliver equitable and higher quality care. This is a pivotal opportunity to showcase how innovation can support human expertise and strengthen trust in healthcare.
AI has the potential to dramatically improve patient outcomes and healthcare cost-efficiency, particularly in high-stakes diagnostic and treatment decisions like oncology, and critical care. In these areas, AI can analyze imaging, lab, and genomic data to uncover patterns that may not be immediately apparent to clinicians. For example, AI tools have shown promise in improving diagnostic accuracy in cancer detection and reducing the time clinicians spend on tasks like charting, allowing for more face-to-face time with patients.
However, these tools must be designed with clinician input at key stages, especially for higher-risk conditions, or tools may be prone to errors or fail to integrate into clinical workflows. By embedding outcome-based contracting (OBC) into federal funding and aligning financial incentives with clinical effectiveness, we are encouraging the development and use of AI tools that have the ability to improve patient outcomes and strengthen the healthcare system’s shift toward value-based care. This supports a broader shift toward value-based care where outcomes, not just outputs, define success.
The connection between OBC and clinician involvement is straightforward. When clinicians are involved in the design and testing of AI tools, these tools are more likely to be effective in real-world settings, thereby improving outcomes and justifying the financial incentives tied to OBC. AI tools can provide significant value for healthcare use in high-stakes, diagnostic and treatment decisions (oncology, cardiology, and critical care) where errors have large consequences on patient outcomes. In those settings, AI can assist by analyzing imaging, lab, and genomic data to uncover patterns that may not be immediately apparent to clinicians. However, these tools should not function autonomously, and input from clinicians is critical to validate AI outputs, specifically for issues where mortality or morbidity is high. In contrast, for lower-risk or routine care of common colds or minor dermatologic conditions, AI may be useful as a time-saving tool that does not require the same depth of clinician oversight.
Plan of Action
These actionable recommendations aim to help federal agencies and health systems embed clinician involvement, representative data, and continuous oversight into the lifecycle of healthcare AI.
Recommendation 1. Federal Agencies Should Require Clinician Involvement in the Development and Testing of AI Tools used in Clinical Settings.
Federal agencies should require clinician involvement in all aspects of the development and testing of AI healthcare tools. This mechanism could be enforced through a combination of agency guidance and tying funding eligibility to specific roles and checkpoints for clinicians. Specifically, agencies like the Office of the National Coordinator for Health Information Technology (ONC), the Food and Drug Administration (FDA) can issue guidance mandating clinician participation, and can tie AI tool development funding to the inclusion of clinicians in the design and testing phases. Guidance can mandate clinician involvement at critical stages for: (1) defining clinical tasks and user interface requirements (2) validating interpretability and performance for diverse populations (3) piloting in real workflows and (4) reviewing for safety and bias metrics. This would ensure AI tools used in clinical settings are human-centered, effective, and safe.
Key stakeholders who may wish to be consulted in this process include offices underneath the Department of Health and Human Services (HHS) such as the Office of the National Coordinator for Health Information Technology (ONC), the Food and Drug Administration (FDA), and the Agency for Healthcare Research and Quality (AHRQ). ONC and FDA should work to issue guidance encouraging clinician engagement during the premarket review. This would allow experts thorough review of scientific data and real-world evidence to ensure that the tools used are human-centered and have the ability to improve the quality of care.
Recommendation 2. Incentivize Clinician Involvement Through Outcomes-Based Contracting
Federal agencies such as the Department of Health and Human Services (HHS), the Centers for Medicare and Medicaid Services (CMS), and the Agency for Healthcare Research and Quality (AHRQ) should incorporate outcomes-based contracting requirements into AI-related healthcare grant programs. Funding should be awarded to grantees who: (1) include clinicians as part of their AI design teams or advisory boards, (2) develop formal clinician feedback loops, and (3) demonstrate measurable outcomes such as improved diagnostic accuracy or workflow efficiency. These outcomes are essential when thinking about clinician engagement and how it will improve the usability of AI tools and their clinical impact.
Key stakeholders include HHS, CMS, ONC, AHRQ, as well as clinicians, AI developers, and potentially patient advocacy organizations. These requirements should prioritize funding for entities that demonstrate clear clinician involvement at key development and testing phases, with metrics tied to improvements in patient outcomes and clinician satisfaction. This model would align with CMS’s ongoing efforts to foster a patient-centered, data-driven healthcare ecosystem that uses tools designed with clinical needs in mind, as recently emphasized during the health tech ecosystem initiative meeting. Embedding outcomes-based contracting into the federal grant process will link funding to clinical effectiveness and incentivize developers to work alongside clinicians through the lifecycle of their AI tools.
Recommendation 3. Develop Standards for AI Interoperability
ONC should develop interoperability guidelines that enable AI systems to share information across platforms while simultaneously protecting patient privacy. As the challenge of healthcare data fragmentation has become evident, AI tools must seamlessly integrate with diverse electronic healthcare records (EHRs) and other clinical platforms to ensure their effectiveness.
An example of successful interoperability frameworks can be seen through the Trusted Exchange Framework and Common Agreement (TEFCA), which aims to establish a nationwide interoperability infrastructure for the exchange of health information. Using a model such as this one can establish seamless integration across different healthcare settings and EHR systems, ultimately promoting efficient and accurate patient care. This effort would involve the consultation of clinicians, electronic health record vendors, patients, and AI developers. These guidelines will help ensure that AI tools can be used safely and effectively across clinical settings.
Recommendation 4. Establish Post-Market Surveillance and Evaluation of Healthcare AI Tools to Enhance Performance and Reliability
Federal agencies such as FDA and AHRQ should establish frameworks that can be used for the continuous monitoring of AI tools in clinical settings. These frameworks for privacy-protected data collection should incorporate feedback loops that allow real-world data from clinicians and patients to inform ongoing updates and improvements to the systems. This ensures the effectiveness and accuracy of the tools over time. Special emphasis should be placed on bias audits that can detect disparities in the system’s performance across different patient groups. Bias audits will be key to identifying whether AI tools inadvertently present disadvantages to specific populations based on the data they were trained on. Agencies should require that these audits be conducted routinely as part of the post-market surveillance process. The surveillance data collected can be used for future development cycles where AI tools are updated or re-trained to address shortcomings.
Evaluation methods should track clinician satisfaction, error rates, diagnostic accuracy, and reportability of failures. During this ongoing evaluation process, incorporating routine bias audits into post-market surveillance will ensure that these tools remain equitable and effective over time. Funding for this initiative could potentially be provided through a zero-cost, fee-based structure or federally appropriated grants. Key stakeholders in this process could include clinicians, AI developers, and patients, all of whom would be responsible for providing oversight.
Conclusion
Integrating AI tools into healthcare has an immense amount of potential to improve patient outcomes, streamline clinical workflows, and reduce errors and bias. However, without clinician involvement in the development and testing of these tools, we risk continual system degradation and patient harm. Requiring that all AI systems used for healthcare are human-centered through clinician input will ensure these systems are effective, safe, and align with real-world clinical needs. This human-centered approach is critical not only for usability, but also for building trust among clinicians and patients, fostering the adoption of AI tools, and ensuring they function properly in real-world clinical settings.
In addition, aligning funding and clinical outcomes through outcomes-based contracting adds a mechanism that forces accountability and ensures lasting impact. When developers are rewarded for improving safety, usability, and equity through clinician involvement, we can transform AI tools into safer care. There is an urgency to address these challenges due to the rapid adoption of AI tools which will require safeguards and ethical oversight. By embedding these recommendations into funding opportunities, we will move America toward building trustworthy healthcare systems that enhance patient safety, clinician expertise, and are adaptive while maximizing AI’s potential for improving patient outcomes. Clinician engagement, both in the development process and through ongoing feedback loops will be the foundation of this transformation. With the right structures in place, we can ensure AI becomes a trusted partner in healthcare and not a risk to it.
This memo produced as part of Strengthening Pathways to Disease Prevention and Improved Health Outcomes.
A National Blueprint for Whole Health Transformation
Despite spending over 17% of GDP on health care, Americans live shorter and less healthy lives than their peers in other high-income countries. Rising chronic disease and mental health challenges as well as clinician burnout expose the limits of a system built to treat illness rather than create health. Addressing chronic disease while controlling healthcare costs is a bipartisan goal, the question now is how to achieve this shared goal? A policy window is opening now as Congress debates health care again – and in our view, it’s time for a “whole health” upgrade.
Whole Health is a proven, evidence-based framework that integrates medical care, behavioral health, public health, and community support so that people can live healthier, longer, and more meaningful lives. Pioneered by the Veterans Health Administration, Whole Health offers a redesign to U.S. health and social systems: it organizes how health is created and supported across sectors, shifting power and responsibility from institutions to people and communities. It begins with what matters most to people–their purpose, aspirations, and connections–and aligns prevention, clinical care, and social supports accordingly. Treating Whole Health as a shared public priority would help ensure that every community has the conditions to thrive.
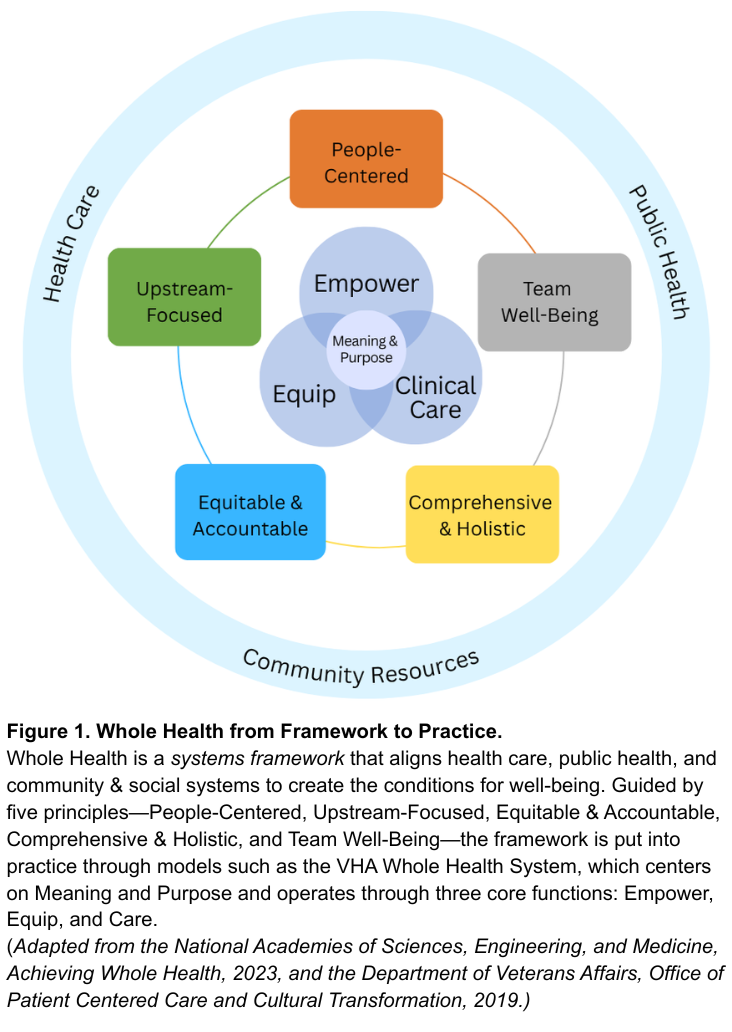
Challenge and Opportunity
The U.S. health system spends over $4 trillion annually, more per capita than any other nation, yet underperforms on life expectancy, infant mortality, and chronic disease management. The prevailing fee-for-service model fragments care across medical, behavioral, and social domains, rewarding treatment over prevention. This fragmentation drives costs upward, fuels clinician burnout, and leaves many communities without coordinated support.
At this inflection point in our declining health outcomes and growing public awareness of the failures of our health system, federal prevention and public health programs are under review, governors are seeking cost-effective chronic disease solutions, and the National Academies is advocating for new healthcare models. Additionally, public demand for evidence-based well-being is growing, with 65% of Americans prioritizing mental and social health. There is clear demand for transformation in our health care system to deliver results in a much more efficient and cost effective way.
Veterans Health Administration’s Whole Health System Debuted in 2011
Whole Health offers a system-wide redesign for the challenge at hand. As defined by the National Academies of Sciences, Engineering, and Medicine, Whole Health is a framework for organizing how health is created and supported across sectors. It integrates medical care, behavioral health, public health, and community resources. As shown in Figure 1, the framework connects five system principles—People-Centered, Upstream-Focused, Equitable & Accountable, Comprehensive & Holistic, and Team Well-Being–that guide implementation across health and social support systems. The nation’s largest health system, the Veterans Health Administration’s (VHA), has demonstrated this framework in clinical practice through their Whole Health System since 2011. The VHA’s Whole Health System operates through three core functions: Empower (helping individuals define purpose), Equip (providing community resources like peer support), and Clinical Care (delivering coordinated, team-based care). Together, these elements align with what matters most to people, shifting the locus of control from expert-driven systems to shared agency through partnerships. The Whole Health System at the VHA has reduced opioid use and improved chronic disease outcomes.
Successful State Examples
Beyond the VHA, states have also demonstrated the possibility and benefits of Whole Health models. North Carolina’s Healthy Opportunities Pilots extended Medicaid coverage to housing, food, and transportation, showing fewer emergency visits and savings of about $85 per member per month. Vermont’s Blueprint for Health links primary care practices with community health teams and social services, reducing expenditures by about $480 per person annually and boosting preventive screenings. Finally, the Program of All-Inclusive Care for the Elderly (PACE), currently being implemented in 33 states, utilizes both Medicare and Medicaid funding to coordinate medical and social care for older adults with complex medical needs. While improvements can be made to national program-wide evaluation, states like Kansas have done evaluations that have found that the PACE program is less expensive than nursing homes per beneficiary and that nursing home admissions decline by 5% to 15% for beneficiaries.
Success across each of these examples relies on three pillars: (1) integrating medical, behavioral, social, and public health resources; (2) sustainable financing that prioritizes prevention and coordination; and (3) rigorous evaluation of outcomes that matter to people and communities. While these programs are early signs of success of Whole Health models, without coordinated leadership, efforts will fragment into isolated pilots and it will be challenging to learn and evolve.
A policy window for rethinking the health care system is opening. At this national inflection point, the U.S. can work to build a unified Whole Health strategy that enables a more effective, affordable and resilient health system.
Plan of Action
To act on this opportunity, federal and state leaders can take the following coordinated actions to embed Whole Health as a unifying framework across health, social, and wellbeing systems.
Recommendation 1. Declare Whole Health a Federal and State Priority.
Whole Health should become a unifying value across federal and state government action on health and wellbeing, embedding prevention, connection, and integration into how health and social systems are organized, financed, and delivered. Actions include:
- Federal Executive Action. The Executive Office of the President should create a Whole Health Strategic Council that brings together Veterans Affairs (VA), Health and Human Services (e.g. Centers for Disease Control and Prevention, Centers for Medicare and Medicaid (CMS), and Health Resources and Services Administration (HRSA)), Housing and Urban Development (HUD), and the U.S. Department of Agriculture (USDA) to align strategies, budgets, and programs with Whole Health principles through cross-agency guidance and joint planning. This council should also work with Governors to establish evidence-based benchmarks for Whole Health operations and evaluation (e.g., person-centered planning, peer support, team integration) and shared outcome metrics for well-being and population health.
- U.S. Congressional Action. Authorize whole health benefits, like housing assistance, nutrition counseling, transportation to appointments, peer support programs, and well-being centers as reimbursable services under Medicare, Medicaid and the Affordable Care Act health subsidies.
- State Action. Adopt Whole Health models through Medicaid managed-care contracts and through CDC and HRSA grant implementation. States should also develop support for Whole Health services in trusted local settings such as libraries, faith-based organizations, senior centers, to reach people where they live and gather.
Recommendation 2. Realign Financing and Payment to Reward Prevention and Team-Based Care.
Federal payment modalities need to shift from a fee-for-service model toward hybrid value-based models. Models such as per-member-per-month payments with quality incentives, can sustain comprehensive, team-based care while delivering outcomes that matter, like reductions in chronic disease and overall perceived wellbeing. Actions include:
- Federal Executive Action. Expand Advanced Primary Care Management (APCM) payments to include Whole Health teams, including clinicians, peer coaches, and community health workers. Ensure that this funding supports coordination, person-centered planning, and upstream prevention, such as food as medicine programs. Further, CMS can expand reimbursements to community health workers and peer support roles and standardize their scope-of-practice rules across states.
- U.S. Congressional Action. Invest in Medicare and Medicaid innovation programs, such as the CMS Innovation Center (CMMI), that reward prevention and chronic disease reduction. Additionally, expand tools for payment flexibility, through Medicaid waivers and state innovation funds, to help states adapt Whole Health models to local needs.
- State Action. Require Medicaid managed-care contracts to reimburse Whole Health services, particularly in underserved and rural areas, and encourage payers to align benefit designs and performance measures around well-being. States should also leverage their state insurance departments to guide and incentivize private health insurers to adopt Whole Health payment models.
Recommendation 3. Strengthen and Expand the Whole Health Workforce.
Whole Health practice needs a broad team to be successful: clinicians, community health workers, peer coaches, community organizations, nutritionists, and educators. To build this workforce, governments need to modernize training, assess the workforce and workplace quality, and connect the fast-growing well-being sector with health and community systems. Actions include:
- Federal Executive Action. Through VA and HRSA establish Whole Health Workforce Centers of Excellence to develop national curricula, set standards, and disseminate evidence on effective Whole Health team-building. Further, CMS should track workforce outcomes such as retention, burnout, and team integration, and evaluate the benefits for health professionals working in Whole Health systems versus traditional health systems.
- U.S. Congressional Action. Expand CMS Graduate Medical Education Funds and HRSA workforce programs to support Whole Health training, certifications, and placements across clinical and community settings.
- State Action. As a part of initiatives to grow the health workforce, state governments should expand the definition of a “health professional” to include Whole Health practitioners. Further, states can leverage their role as a licensure for professionals by creating a “whole health” licensing process that recognizes professionals that meet evidence-based standards for Whole Health.
Recommendation 4. Build a National Learning and Research Infrastructure.
Whole Health programs across the country are proving effective, but lessons remain siloed. A coordinated national system should link evidence, evaluation, and implementation so that successful models can scale quickly and sustainably.
- Federal Executive Action. Direct the Agency for Healthcare Research and Quality, National Institutes of Health, and partner agencies (VA, HUD, USDA) to run pragmatic trials and cost-effectiveness studies of Whole Health interventions that measure well-being across clinical, biomedical, behavioral, and social domains. The federal government should also embed Whole Health frameworks into government-wide research agendas to sustain a culture of evidence-based improvement.
- U.S. Congressional Action. Charter a quasi-governmental entity, modeled on Patient-Centered Outcomes Research Institute (PCORI), to coordinate Whole Health demonstration sites and research. This new entity should partner with CMMI, HRSA and VA to test Whole Health payment and delivery models under real-world conditions. This entity should also establish an interagency team as well as state network to address payment, regulatory, and privacy barriers identified by sites and pilots.
- State Action. Partner with federal agencies through innovation waivers (e.g. 1115 waivers and 1332 waivers) and learning collaboratives to test Whole Health models and share data across state systems and with the federal government.
Conclusion
The United States spends more on health care than any other nation yet delivers poorer outcomes. Whole Health offers a proven path to reverse this trend, reframing care around prevention, purpose, and integration across health and social systems. Embedding Whole Health as the operating system for America’s health requires three shifts: (1) redefining the purpose from treating disease to optimizing health and well-being; (2) restructuring care to empower, equip, and treat through team-based and community-linked approaches; and (3) rebalancing control from expert-driven systems to partnerships guided by what matters most to people and communities. Federal and state leaders have the opportunity to turn scattered Whole Health pilots to a coordinated national strategy. The cost of inaction is continued fragmentation; the reward of action is a healthier and more resilient nation.
This memo produced as part of Strengthening Pathways to Disease Prevention and Improved Health Outcomes.
Both approaches emphasize caring for people as integrated beings rather than as a collection of diseases, but they differ in scope and application. Whole Person Health, as used by NIH, focuses on the biological, psychological, and behavioral systems within an individual—it is primarily a research framework for understanding health across body systems. Whole Health is a systems framework that extends beyond the individual to include families, communities, and environments. It integrates medical care, behavioral health, public health, and social support around what matters most to each person. In short, Whole Person Health is about how the body and mind work together; Whole Health is about how health, social, and community systems work together to create the conditions for well-being. Policymakers can use Whole Health to guide financing, workforce, and infrastructure reforms that translate Whole Person Health science into everyday practice.
Integrative Health combines evidence-based conventional and complementary approaches such as mindfulness, acupuncture, yoga, and nutrition to support healing of the whole person. Whole Health extends further. It includes prevention, self-care, and personal agency, and moves beyond the clinic to connect medical care with social, behavioral, and community dimensions of health. Whole Health uses integrative approaches when evidence supports them, but it is ultimately a systems model that aligns health, social, and community supports around what matters most to people. For policymakers, it provides a structure for integrating clinical and community services within financing and workforce strategies.
They share a common foundation but differ in scope and audience. The VA Whole Health System, developed by the Department of Veterans Affairs, is an operational model, a way of delivering care that helps veterans identify what matters most, supports self-care and skill building, and provides team-based clinical treatment. The National Academies’ Whole Health framework builds on the VA’s experience and expands it to the national level. It is a policy and systems framework that applies Whole Health principles across all populations and connects health care with public health, behavioral health, and community systems. In short, the VA model shows how Whole Health works in practice, while the National Academies framework shows how it can guide national policy and system alignment.
In Honor of Patient Safety Day, Four Recommendations to Improve Healthcare Outcomes
Through partnership with the Doris Duke Foundation, FAS is working to ensure that rigorous, evidence-based ideas on the cutting edge of disease prevention and health outcomes are reaching decision makers in an effective and timely manner. To that end, we have been collaborating with the Strengthening Pathways effort, a series of national conversations held in spring 2025 to surface research questions, incentives, and overlooked opportunities for innovation with potential to prevent disease and improve outcomes of care in the United States. FAS is leveraging its skills in policy entrepreneurship, working with session organizers, to ensure that ideas surfaced in these symposia reach decision-makers to drive impact in active policy windows.
On this World Patient Safety Day 2025, we share a set of recommendations that align with the National Quality Strategy of Centers for Medicare and Medicaid Services (CMS) goal for zero preventable harm in healthcare. Working with Patients for Patient Safety US, which co-led one of Strengthening Pathways conversations this spring with the Johns Hopkins University Armstrong Institute for Patient Safety and Quality, the issue brief below outlines a bold, modernized approach that uses Artificial Intelligence technology to empower patients and drive change. FAS continues to explore the rapidly evolving AI and healthcare nexus.
Patient safety is an often-overlooked challenge in our healthcare systems. Whether safety events are caused by medical error, missed or delayed diagnoses, deviations from standards of care, or neglect, hundreds of billions of dollars and hundreds of thousands of lives are lost each year due to patient safety lapses in our healthcare settings. But most patient safety challenges are not really captured and there are not enough tools to empower clinicians to improve. Here we present four critical proposals for improving patient safety that are worthy of attention and action.
Challenge and Opportunity
Reducing patient death and harm from medical error surfaced as a U.S. public health priority at the turn of the century with the landmark National Academy of Sciences (NAS) report, To Err is Human: Building a Safer Health System (2000). Research shows that medical error is the 3rd largest cause of preventable death in the U.S. Analysis of Medicare claims data and electronic health records by the Department of Health and Human Services (DHHS) Office of the Inspector General (OIG) in a series of reports from 2008 to 2025 consistently finds that 25-30% of Medicare recipients experience harm events across multiple healthcare settings, from hospitals to skilled nursing facilities to long term care hospitals to rehab centers. Research on the broader population finds similar rates for adult patients in hospitals. The most recent study on preventable harm in ambulatory care found that 7% of patients experienced at least one adverse event, with wide variation of 1.8% to 23.6% from clinical setting to clinical setting. Improving diagnostic safety has emerged as the largest opportunity for patient harm prevention. New research estimates 795,000 patients in the U.S. annually experience death or harm due to missed, delayed or ineffectively communicated diagnoses. The annual cost to the health care system of preventable harm and its health care cascades is conservatively estimated to exceed $200 billion. This cost is ultimately borne by families and taxpayers.
In its National Quality Strategy, the Centers for Medicare and Medicaid Services (CMS) articulated an aspirational goal of zero preventable harm in healthcare. The National Action Alliance for Patient and Workforce Safety, now managed by the Agency for Healthcare Research and Quality (AHRQ), has a goal of 50% reduction in preventable harm by 2026. These goals cannot be achieved without a bold, modernized approach that uses AI technology to empower patients and drive change. Under-reporting negative outcomes and patient harms keeps clinicians and staff from identifying and implementing solutions to improve care. In its latest analysis (July 2025), the OIG finds that fewer than 5% of medical errors are ever reported to the systems designed to gather insights from them. Hospitals failed to capture half of harm events identified via medical record review, and even among captured events, few led to investigation or safety improvements. Only 16% of events required to be reported externally to CMS or State entities were actually reported, meaning critical oversight systems are missing safety signals entirely.
Multiple research papers over the last 20 years find that patients will report things that providers do not. But there has been no simple, trusted way for patient observations to reach the right people at the right time in a way that supports learning and Improvement. Patients could be especially effective in reporting missed or delayed diagnoses, which often manifest across the continuum of care, not in one healthcare setting or a single patient visit. The advent of AI systems provides an unprecedented opportunity to address patient safety and improve patient outcomes if we can improve the data available on the frequency and nature of medical errors. Here we present four ideas for improving patient safety.
Recommendation 1. Create AI-Empowered Safety Event Reporting and Learning System With and For Patients
The Department of Health and Human Services (HHS) can, through CMS, AHRQ or another HHS agency, develop an AI-empowered National Patient Safety Learning and Reporting System that enables anyone, including patients and families, to directly report harm events or flag safety concerns for improvement, including in real or near real time. Doing so would make sure everyone in the system has the full picture — so healthcare providers can act quickly, learn faster, and protect more patients.
This system will:
- Develop a reporting portal to collect, triage and analyze patient reported data directly from beneficiaries to improve patient and diagnostic safety.
- Redesign and modernize Consumer Assessment of Healthcare Providers and Systems
(CAHPS) surveys to include questions that capture beneficiaries’ experiences and outcomes related to patient and diagnostic safety events.
- Redefine the Beneficiary and Family Centered Care Quality Improvement Organizations (BFCC QIO) scope of work to integrate the QIOs into the National Patient Safety Learning and Reporting System.
The learning system will:
- Use advanced triage (including AI) to distinguish high-signal events and route credible
reports directly to the care team and oversight bodies that can act on them.
- Solicit timely feedback and insights in support of hospitals, clinics, and nursing homes to prevent recurrence, as well as feedback over time on patient outcomes that manifest later, e.g. as a result of missed or delayed diagnoses.
- Protect patients and providers by focusing on efficacy of solutions, not blame assignment.
- Feed anonymized, interoperable data into a national learning network that will spot systemic risks sooner and make aggregated data available for transparency and system learning.
Recommendation 2. Create a Real-time ‘Patient Safety Dashboard’ using AI
HHS should build an AI-driven platform that integrates patient-reported safety data — including data from the new National Patient Reporting and Learning System, recommended above — with clinical data from electronic health records to create a real-time ‘patient safety dashboard’ for hospitals and clinics. This dashboard will empower providers to improve care in real time, and will:
- Assist health care providers make accurate and timely diagnoses and avoid errors.
- Make patient reporting easy, effective, and actionable.
- Use AI to triage harm signals and detect systemic risk in real time.
- Build shared national infrastructure for healthcare reporting for all stakeholders.
- Align incentives to reward harm reduction and safety.
By harnessing the power of AI providers will be able to respond faster, identify patients at risk more effectively, and prevent harm thereby improving outcomes. This “central nervous system” for patient safety will be deployed nationally to help detect safety signals in real time, connect information across settings, and alert teams before harm occurs.
Recommendation 3. Mine Billing Data for Deviations from Standards of Care
Standards of care are guidelines that define the process, procedures and treatments that patients should receive in various medical and professional contexts. Standards ensure that individuals receive appropriate and effective care based on established practices. Most standards of care are developed and promulgated by medical societies. But not all clinicians and clinical settings adhere to standards of care, and deviations from standards of care are normal depending upon the case before them. Nonetheless, standards of care exist for a reason and deviations from standards of care should be noted when medical errors result in negative outcomes for patients so that clinicians can learn from these outcomes and improve.
Some patient safety challenges are evident right in the billing data submitted to CMS and insurers. For example, deviations from standards of care can be captured in billing data by comparing clinical diagnosis codes with billing codes and then compared to widely accepted standards of care. By using CMS billing data, the government could identify opportunities for driving the development, augmentation, and wider adoption of standards of care by showing variability and compliance with standards of care for patients, reducing medical error and improving outcomes.
Giving standard setters real data to adapt and develop new standards of care is a powerful tool for improving patient outcomes.
Recommendation 4. Create a Patient Safety AI Testbed
HHS can also establish a Patient Safety AI Testbed to evaluate how AI tools used in diagnosis, monitoring, and care coordination perform in real-world settings. This testbed will ensure that AI improves safety, not just efficiency — and can be co-led by patients, clinicians, and independent safety experts. This is an expansion of the testbeds in the HHS AI Strategic Plan.
The Patient Safety Testbed could include:
- Funding for independent AI test environments to monitor real-world safety and performance over time.
- Public reliability benchmarks and “AI safety labeling”.
- Required participation by AI vendors and provider systems.
Conclusion
There are several key steps that the government can take to address the major loss of health, dollars, and lives due to medical errors, while simultaneously bolstering treatment guidelines, driving the development of new transparent data, and holding the medical establishment accountable for improving care. Here we present four proposals. None of them are particularly expensive when juxtaposed against the tremendous savings they will drive throughout our healthcare system. We can only hope that the Administration’s commitment to patient safety is such that they will adopt them and drive a new era where caregivers, healthcare systems and insurance payers work together to improve patient safety and care standards.
This memo produced as part of Strengthening Pathways to Disease Prevention and Improved Health Outcomes.
Protecting Infant Nutrition Security:
Shifting the Paradigm on Breastfeeding to Build a Healthier Future for all Americans
The health and wellbeing of American babies have been put at risk in recent years, and we can do better. Recent events have revealed deep vulnerabilities in our nation’s infant nutritional security. For example: Pandemic-induced disruptions in maternity care practices that support the establishment of breastfeeding; the infant formula recall and resulting shortage; and a spate of weather-related natural disasters have demonstrated infrastructure gaps and a lack of resilience to safety and supply chain challenges. All put babies in danger during times of crisis.
Breastfeeding is foundational to lifelong health and wellness, but systemic barriers prevent many families from meeting their breastfeeding goals. The policies and infrastructure surrounding postpartum families often limit their ability to succeed in breastfeeding. Despite important benefits, new data from the CDC shows that while 84.1% of infants start out breastfeeding, these numbers fall dramatically in the weeks after birth, with only 57.5% of infants breastfeeding exclusively at one month of age. Disparities persist across geographic location, and other sociodemographic factors, including race/ethnicity, maternal age, and education. Breastfeeding rates in North America are the lowest in the world. Longstanding evidence shows that it is not a lack of desire but rather a lack of support, access, and resources that creates these barriers.
This administration has an opportunity to take a systems approach to increasing support for breastfeeding and making parenting easier for new mothers. Key policy changes to address systemic barriers include providing guidance to states on expanding Medicaid coverage of donor milk, building breastfeeding support and protection into the existing emergency response framework at the Federal Emergency Management Agency, and expressing support for establishing a national paid leave program.
Policymakers on both sides of the aisle agree that no baby should ever go hungry, as evidenced by the bipartisan passage of recent breastfeeding legislation (detailed below) and widely supported regulations. However, significant barriers remain. This administration has the power to address long-standing inequities and set the stage for the next generation of parents and infants to thrive. Ensuring that every family has the support they need to make the best decisions for their child’s health and wellness benefits the individual, the family, the community, and the economy.
Challenge and Opportunity
Breastfeeding plays an essential role in establishing good nutrition and healthy weight, reducing the risk of chronic disease and infant mortality, and improving maternal and infant health outcomes. Breastfed children have a decreased risk of obesity, type 1 and 2 diabetes, asthma, and childhood leukemia. Women who breastfeed reduce their risk of specific chronic diseases, including type 2 diabetes, cardiovascular disease, and breast and ovarian cancers. On a relational level, the hormones produced while breastfeeding, like oxytocin, enhance the maternal-infant bond and emotional well-being. The American Academy of Pediatrics recommends infants be exclusively breastfed for approximately six months with continued breastfeeding while introducing complementary foods for two years or as long as mutually desired by the mother and child.
Despite the well-documented health benefits of breastfeeding, deep inequities in healthcare, community, and employment settings impede success. Systemic barriers disproportionately impact Black, Indigenous, and other communities of color, as well as families in rural and economically distressed areas. These populations already bear the weight of numerous health inequities, including limited access to nutritious foods and higher rates of chronic disease—issues that breastfeeding could help mitigate.
Breastfeeding Saves Dollars and Makes Sense
Low breastfeeding rates in the United States cost our nation millions of dollars through higher health system costs, lost productivity, and higher household expenditures. Not breastfeeding is associated with economic losses of about $302 billion annually or 0.49% of world gross national income. At the national level, improving breastfeeding practices through programs and policies is one of the best investments a country can make, as every dollar invested is estimated to result in a $35 economic return.
In the United States, chronic disease management results in trillions of dollars in annual healthcare costs, which increased breastfeeding rates could help reduce. In the workplace setting, employers see significant cost savings when their workers are able to maintain breastfeeding after returning to work. Increased breastfeeding rates are also associated with reduced environmental impact and associated expenses. Savings can be seen at home as well, as following optimal breastfeeding practices reduces household expenditures. Investments in infant nutrition last a lifetime, paying long-term dividends critical for economic and human development. Economists have completed cost-benefit analyses, finding that investments in nutrition are one of the best value-for-money development actions, laying the groundwork for the success of investments in other sectors.
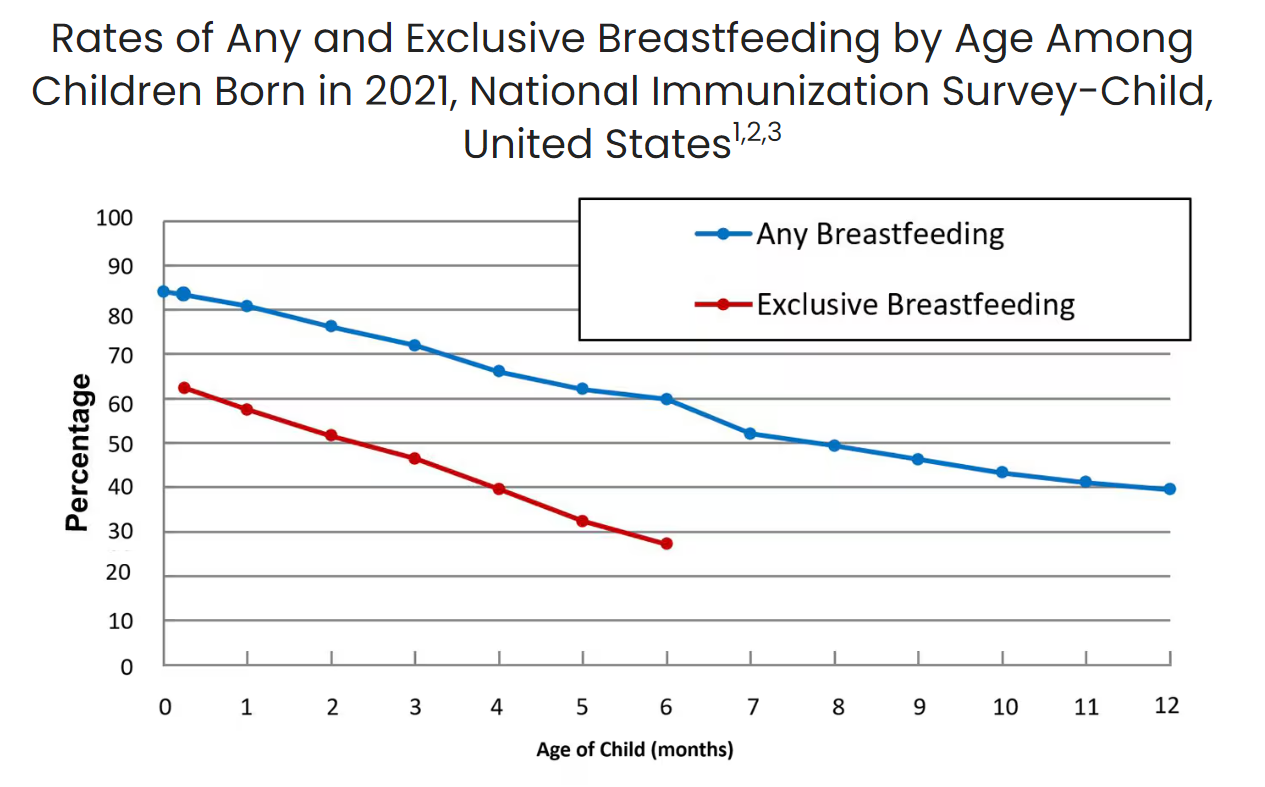
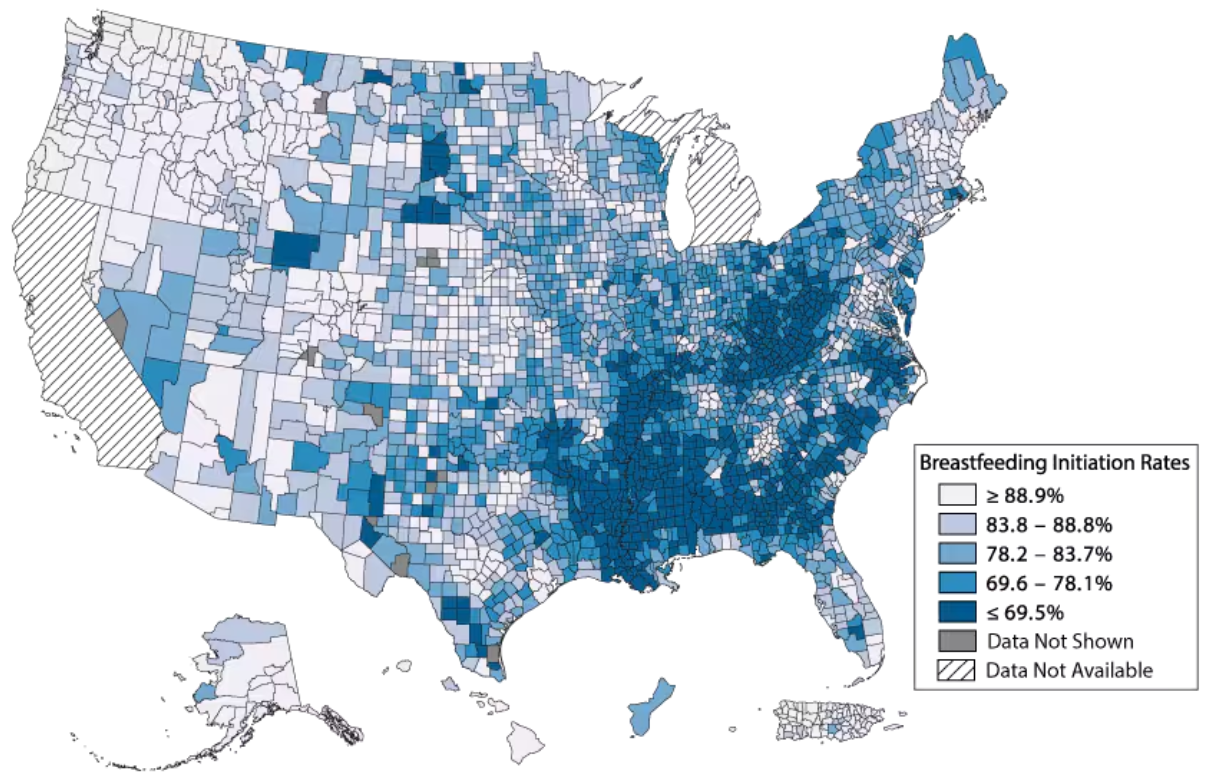
Ongoing trends in breastfeeding outcomes indicate that there are entrenched policy-level challenges and barriers that need to be addressed to ensure that all infants have an opportunity to benefit from access to human milk. Currently, for too many families, the odds are stacked against them. It’s not a question of individual choice but one of systemic injustice. Families are often forced into feeding decisions that do not reflect their true desires due to a lack of accessible resources, support, and infrastructure.
While the current landscape is rife with challenges, the solutions are known and the potential benefits are tremendous. This administration has the opportunity to realize these benefits and implement a smart and strategic response to the urgent situation that our nation is facing just as the political will is at an all-time high.
The History of Breastfeeding Policy
In the late 1960s and early 1970s less than 30 percent of infants were breastfed. The concerted efforts of individuals and organizations and the emergence of the field of lactation have worked to counteract or remove many barriers, and policymakers have sent a clear and consistent message that breastfeeding is bipartisan. This is evident in the range of recent lactation-friendly legislation, including:
- Bottles and Breastfeeding Equipment Screening Act (BABES Act) in 2016
- Friendly Airports for Mothers (FAM) Act in 2018
- Fairness for Breastfeeding Mothers Act in 2019
- Providing Urgent Maternal Protections (PUMP) for Nursing Mothers Act in 2022
Administrative efforts ranging from the Business Case for Breastfeeding to The Surgeon General’s Call to Action to Support Breastfeeding and the armed services updates on uniform requirements for lactating soldiers demonstrate a clear commitment to breastfeeding support across the decades.
These policy changes have made a difference. But additional attention and investment, with a particular focus on the birth and early postpartum period, as well as during and after emergencies, is needed to secure the potential health and economic benefits of comprehensive societal support for breastfeeding. This administration can take considerable steps toward improving U.S. health and wellness and protecting infant nutrition security.
Plan of Action
A range of federal agencies coordinate programs, services, and initiatives impacting the breastfeeding journey for new parents. Expanding and building on existing efforts through the following steps can help address some of today’s most pressing barriers to breastfeeding.
Each of the recommended actions can be implemented independently and would create meaningful, incremental change for families. However, a comprehensive approach that implements all these recommendations would create the marked shift in the landscape needed to improve breastfeeding initiation and duration rates and establish this administration as a champion for breastfeeding families.
Recommendation 1. Increase access to pasteurized donor human milk by directing the Centers for Medicare & Medicaid Services (CMS) to provide guidance to states on expanding Medicaid coverage.
Pasteurized donor human milk is lifesaving for vulnerable infants, particularly those born preterm or with serious health complications. Across the United States, milk banks gently pasteurize donated human milk and distribute it to fragile infants in need. This lifesaving liquid gold reduces mortality rates, lowers healthcare costs, and shortens hospital stays. Specifically, the use of donor milk is associated with increased survival rates and lowered rates of infections, sepsis, serious lung disease, and gastrointestinal complications. In 2022, there were 380,548 preterm births in the United States, representing 10.4% of live births, so the potential for health and cost savings is substantial. Data from one study shows that the cost of a neonatal intensive care unit stay for infants at very low birth weight is nearly $220,000 for 56 days. The use of donor human milk can reduce hospital length of stay by 18-50 days by preventing the development of necrotizing enterocolitis in preterm infants. The benefits of human milk extend beyond the inpatient stay, with infants receiving all human milk diets in the NICU experiencing fewer hospital readmissions and better overall long-term outcomes.
Although donor milk has important health implications for vulnerable infants in all communities and can result in significant economic benefit, donor milk is not equitably accessible. While milk banks serve all states, not all communities have easy access to donated human milk. Moreover, many insurers are not required to cover the cost, creating significant barriers to access and contributing to racial and geographic disparities.
To ensure that more babies in need have access to lifesaving donor milk, the administration should work with CMS to expand donor milk coverage under state Medicaid programs. Medicaid covers approximately 40% of all US births and 50% of all early preterm births. Medicaid programs in at least 17 states and the District of Columbia already include coverage of donor milk. The administration can expand access to this precious milk, help reduce health care costs, and address racial and geographic disparities by releasing guidance for the remaining states regarding coverage options in Medicaid.
Recommendation 2. Include infant feeding in Federal Emergency Management Agency (FEMA) emergency planning and response.
Infants and children are among the most vulnerable in an emergency, so it is critical that their unique needs are considered and included in emergency planning and response guidance. Breastfeeding provides clean, optimal nutrition, requires no fuel, water, or electricity, and is available, even in the direst circumstances. Human milk contains antibodies that fight infection, including diarrhea and respiratory infections common among infants in emergency situations. Yet efforts to protect infant and young child feeding in emergencies are sorely lacking, particularly in the immediate aftermath of disasters and emergencies.
Ensuring access to lactation support and supplies as part of emergency response efforts is essential for protecting the health and safety of infants. Active support and coordination between federal, state, and local governments, the commercial milk formula industry, lactation support providers, and all other relevant actors involved in response to emergencies is needed to ensure safe infant and young child feeding practices and equitable access to support. There are two simple, cost-effective steps that FEMA can take to protect breastfeeding, preserve resources, and thus save additional lives during emergencies.
- Require FEMA to participate in the Federal Interagency Breastfeeding Workgroup, a collection of federal agencies that come together to connect and collaborate on breastfeeding issues, to ensure coordination across agencies.
- Update the FEMA Public Assistance Program and Policy Guide (PAPPG) to include breastfeeding and lactation as a functional need in the section on Congregate Shelter Services so that emergency response efforts can include services from lactation support providers and be reimbursed for the associated costs.
Recommendation 3. Expand access to paid family & medical leave by including paid leave as a priority in the President’s Budget and supporting the efforts of the bipartisan, bicameral congressional Paid Leave Working Group.
Employment policies in the United States make breastfeeding harder than it needs to be. The United States is one of the only countries in the world without a national paid family and medical leave program. Many parents return to work quickly after birth, before a strong breastfeeding relationship is established, because they cannot afford to take unpaid leave or because they do not qualify for paid leave programs with their employer or through state or local programs. Nearly 1 in 4 employed mothers return to work within two weeks of childbirth.
Paid family leave programs make it possible for employees to take time for childbirth recovery, bond with their baby, establish feeding routines, and adjust to life with a new child without threatening their family’s economic well-being. This precious time provides the foundation for success, contributing to improved rates of breastfeeding initiation and duration, yet only a small portion of workers are able to access it. There are significant disparities in access to paid leave among racial and ethnic groups, with Black and Hispanic employees less likely than their white non-Hispanic counterparts to have access to paid parental leave. There are similar disparities in breastfeeding outcomes among racial groups.
The momentum is building substantially to improve the paid family and medical leave landscape in the United States. Thirteen states and the District of Columbia have established mandatory state paid family leave systems. Supporting paid leave has become an important component of candidate campaign plans, and bipartisan support for establishing a national program remains strong among voters. The formation of Bipartisan Paid Family Leave Working Groups in both the House and Senate demonstrate commitment from policymakers on both sides of the aisle.
By directing the Office of Management and Budget to include funding for paid leave in the President’s Budget recommendation and working collaboratively with the Congressional Paid Leave Working Groups, the administration can advance federal efforts to increase access to paid family and medical leave, improving public health and helping American businesses.
Conclusion
These three strategies offer the opportunity for the White House to make an immediate and lasting impact by protecting infant nutrition security and addressing disparities in breastfeeding rates, on day one of the Presidential term. A systems approach that utilizes multiple strategies for integrating breastfeeding into existing programs and efforts would help shift the paradigm for new families by addressing long-standing barriers that disproportionately affect marginalized communities—particularly Black, Indigenous, and families of color. A clear and concerted effort from the Administration, as outlined, offers the opportunity to benefit all families and future generations of American babies.
The administration’s focused and strategic efforts will create a healthier, more supportive world for babies, families, and breastfeeding parents, improve maternal and child health outcomes, and strengthen the economy. This administration has the chance to positively shape the future for generations of American families, ensuring that every baby gets the best possible start in life and that every parent feels empowered and supported.
Now is the time to build on recent momentum and create a world where families have true autonomy in infant feeding decisions. A world where paid family leave allows parents the time to heal, bond, and establish feeding routines; communities provide equitable access to donor milk; and federal, state, and local agencies have formal plans to protect infant feeding during emergencies, ensuring no baby is left vulnerable. Every family deserves to feel empowered and supported in making the best choices for their children, with equitable access to resources and support systems.
This policy memo was written with support from Suzan Ajlouni, Public Health Writing Specialist at the U.S. Breastfeeding Committee. The policy recommendations have been identified through the collective learning, idea sharing, and expertise of USBC members and partners.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
Rather than being a matter of personal choice, infant feeding practice is informed by circumstance and level (or lack) of support. When roadblocks exist at every turn, families are backed into a decision because the alternatives are not available, attainable, or viable. United States policies and infrastructure were not built with the realities of breastfeeding in mind. Change is needed to ensure that all who choose to breastfeed are able to meet their personal breastfeeding goals, and society at large reaps the beneficial social and economic outcomes.
The Fiscal Year 2024 President’s Budget proposed to establish a national, comprehensive paid family and medical leave program, providing up to 12 weeks of leave to allow eligible workers to take time off to care for and bond with a new child; care for a seriously ill loved one; heal from their own serious illness; address circumstances arising from a loved one’s military deployment; or find safety from domestic violence, sexual assault, or stalking. The budget recommendation included $325 billion for this program. It’s important to look at this with the return on investment in mind, including improved labor force attachment and increased earnings for women; better outcomes and reduced health care costs for ill, injured, or disabled loved ones; savings to other tax-funded programs, including Medicaid, SNAP, and other forms of public assistance; and national economic growth, jobs growth, and increased economic activity.
There are a variety of national monitoring and surveillance efforts tracking breastfeeding initiation, duration, and exclusivity rates that will inform how well these actions are working for the American people, including the National Immunization Survey (NIS), Pregnancy Risk Assessment and Monitoring System (PRAMS), Infant Feeding Practices Study, and National Vital Statistics System. The CDC Breastfeeding Report card is published biannually to bring these key data points together and place them into context. Significant improvements in the data have already been seen across recent decades, with breastfeeding initiation rates increasing from 73.1 percent in 2004 to 84.1 percent in 2021.
The U.S. Breastfeeding Committee is a coalition bringing together approximately 140 organizations from coast to coast representing the grassroots to the treetops – including federal agencies, national, state, tribal, and territorial organizations, and for-profit businesses – that support the USBC mission to create a landscape of breastfeeding support across the United States. Nationwide, a network of hundreds of thousands of grassroots advocates from across the political spectrum support efforts like these. Together, we are committed to ensuring that all families in the U.S. have the support, resources, and accommodations to achieve their breastfeeding goals in the communities where they live, learn, work, and play. The U.S. Breastfeeding Committee and our network stand ready to work with the administration to advance this plan of action.
Clearing the Path for New Uses for Generic Drugs
The labeling-only 505(b)(2) NDA pathway for non-manufacturers to seek FDA approval
Repurposing generic drugs as new treatments for life-threatening diseases is an exciting yet largely overlooked opportunity due to a lack of market-driven incentives. The low profit margins for generic drugs mean that pharmaceutical companies rarely invest in research, regulatory efforts, and marketing for new uses. Nonprofit organizations and other non-commercial non-manufacturers are increasing efforts to repurpose widely available generic drugs and rapidly expand affordable treatment options for patients. However, these non-manufacturers find it difficult to obtain regulatory approval in the U.S. They face significant challenges in using the existing approval pathways, specifically in: 1) providing the FDA with required chemistry, manufacturing, and controls (CMC) data, 2) providing the FDA with product samples, and 3) conducting post-marketing surveillance. Without a straightforward path for approval and updating drug labeling, non-manufacturers have relied on off-label use of repurposed drugs to drive uptake. This practice results in outdated labeling for generics and hinders widespread clinical adoption, limiting patient access to these potentially life-saving treatments.
To encourage greater adoption of generic drugs in clinical practice – that is, to encourage the repurposing of these drugs – the FDA should implement a dedicated regulatory pathway for non-manufacturers to seek approval of new indications for repurposed generic drugs. A potential solution is an extension of the existing 505(b)(2) new drug application (NDA) approval pathway. This extension, the “labeling-only” 505(b)(2) NDA, would be a dedicated pathway for non-manufacturers to seek FDA approval of new indications for well-established small molecule drugs when multiple generic products are already available. The labeling-only 505(b)(2) pathway would be applicable for repurposing drugs for any disease. Creating a regulatory pathway for non-manufacturers would unlock access to innovative therapies and enable the public to benefit from the enormous potential of low-cost generic drugs.
Challenge and Opportunity
The opportunity for generic drug repurposing
On-patent, branded drugs are often unaffordable for Americans. Due to the high cost of care, 42% of patients in the U.S. exhaust their life savings within two years of a cancer diagnosis. Generic drug repurposing – the process of finding new uses for FDA-approved generic drugs – is a major opportunity to quickly create low-cost and accessible treatment options for many diseases. In oncology, hundreds of generic drugs approved for non-cancer uses have been tested as cancer treatments in published preclinical and clinical studies.
The untapped potential for generic drug repurposing in cancer and other diseases is not being realized because of the lack of market incentives. Pharmaceutical companies are primarily focused on de novo drug development to create new molecular and chemical entities. Typically, pharmaceutical companies will invest in repurposing only when the drugs are protected by patents or statutory market exclusivities, or when modification to the drugs can create new patent protection or exclusivities (e.g., through new formulations, dosage forms, or routes of administration). Once patents and exclusivities expire, the introduction of generic drugs creates competition in the marketplace. Generics can be up to 80-85% less expensive than their branded counterparts, driving down overall drug prices. The steep decline in prices means that pharmaceutical companies have little motivation to invest in research and marketing for new uses of off-patent drugs, and this loss of interest often starts in the years preceding generic entry.
In theory, pharmaceutical companies could repurpose generics without changing the drugs and apply for method-of-use patents, which should provide exclusivity for new indications and the potential for higher pricing. However, due to substitution of generic drugs at the pharmacy level, method-of-use patents are of little to no practical value when there are already therapeutically equivalent products on the market. Pharmacists can dispense a generic version instead of the patent-protected drug product, even if the substituted generic does not have the specific indication in its labeling. Currently, nearly all U.S. states permit substitution by the pharmacy, and over a third have regulations that require generic substitution when available.
Nonprofits like Reboot Rx and other non-commercial non-manufacturers are therefore stepping in to advance the use of repurposed generic drugs across many diseases. Non-manufacturers, which do not manufacture or distribute drugs, aim to ensure there is substantial evidence for new indications of generic drugs and then advocate for their clinical use. Regulatory approval would accelerate adoption. However, even with substantial evidence to support regulatory review, non-manufacturers find it difficult or impossible to seek approval for new indications of generic drugs. There is no straightforward pathway to do so within the current U.S. regulatory framework without offering a specific, manufactured version of the drug. This challenge is not unique to the U.S.; recent efforts in the European Union (EU) have sought to address the regulatory gap. In the 2023 EU reform of pharmaceutical legislation, Article 48 is currently under review by the European Parliament as a potential solution to allow nonprofit entities to spearhead submissions for the approval of new indications for authorized medicinal products with the European Medicines Agency. To maximize the patient impact of generic drugs in America, non-manufacturers should be able to drive updates to FDA drug labeling, enabling widespread clinical adoption of repurposed drugs in a formal, predictable, and systematic manner.
The importance of FDA approval
Drugs that are FDA-approved can be prescribed for any indication not listed on the product labeling, often referred to as “off-label use”. Since non-manufacturers face significant challenges pursuing regulatory approval for new indications, they often must rely on advocating for off-label use of repurposed drugs.
While off-label use is widely accepted and helpful for specific circumstances, there are significant advantages to having FDA approval of new drug indications included in labeling. FDA drug labeling is intended to contain up-to-date information about drug products and ensures that the necessary conditions of use (including dosing, warnings, and precautions) are communicated for the specific indications. It is the primary authoritative source for making informed treatment decisions and is heavily valued by the medical community. Approval may increase the likelihood of uptake by clinical guidelines, pathways systems, and healthcare payers. Indications with FDA approval may generate greater awareness of the treatment options, leading to a broader and more rapid impact on clinical practice.
Clinical practice guidelines are often the leading authority for prescribers and patients regarding off-label use. In oncology, for example, the National Comprehensive Cancer Network (NCCN) Guidelines are commonly used guidelines that include many off-label uses. However, guidelines do not exist for every disease and medical specialty, which can make it more difficult to gain acceptance for off-label uses. The Centers for Medicare and Medicaid Services (CMS) policy routinely covers off-label drug uses if they are listed in certain compendia. The NCCN Compendia, which is based on the NCCN Guidelines, is the only accepted compendium that is disease-specific.
Off-label use requires more effort from individual prescribers and patients to independently evaluate new drug data, thereby slowing uptake of the treatments. This can be especially difficult for community-based physicians, who need to remain up-to-date on new treatment options across many diseases. Off-label prescribing can also introduce medico-legal risks, such as malpractice. These burdens and risks limit off-label prescribing, even when there is supportive evidence for the new uses.
As new uses for generic drugs are discovered, it is crucial to update the labeling to ensure alignment with current clinical practice. Outdated generic drug labeling means that prescribers and patients may not have access to all the necessary information to understand the full risk-benefit profile. Americans deserve to have access to all effective treatment options – especially low-cost and widely available generic drugs that could help mitigate the financial toxicity and health inequities faced by many patients. For the public benefit, the FDA should support approaches that remove regulatory barriers for non-manufacturers and modernize drug labeling.
Existing pathways for manufacturers to obtain FDA approval
The current FDA approval system is based on the idea that sponsors have discrete physical drug products. Traditionally, sponsors seeking FDA approval are pharmaceutical companies or drug manufacturers that intend to produce (or contract for production), distribute, and sell the finished drug product. For the purposes of FDA regulation, “drug” refers to a substance intended for use in the treatment or prevention of disease; “drug product” is the final dosage form that contains a drug substance and inactive ingredients made and sold by a specific manufacturer. One drug can be present on the market in multiple drug products. In the current regulatory framework, drug products are approved through one of the following:
- 505(b)(1) NDA: New drug products, including new molecular entities, are first submitted for FDA review through the 505(b)(1) new drug application (NDA) pathway. In this pathway, the sponsor either generates all necessary clinical data or owns the right of reference to the clinical data needed to support the NDA. FDA approval of an NDA establishes a reference listed drug (RLD), based on the FDA’s determinations of safety and effectiveness for the drug.
- 505(b)(2) NDA: For new drug products or formulations utilizing or related to previously approved drugs or drug substances, the 505(b)(2) NDA pathway offers a streamlined alternative to the 505(b)(1) NDA. By allowing the sponsor to reference the FDA’s findings of safety and effectiveness for previously approved drugs and published literature, the sponsor can submit data they do not own or for which they do not have the right of reference, once any exclusivities expire.
- ANDA: Generic drug equivalents are approved through the 505(j) abbreviated NDA (ANDA) pathway. The RLD serves as the standard that other manufacturers may reference when seeking approval for generic versions of the drug. Upon approval, ANDA products receive a therapeutic equivalence designation in the FDA’s Orange Book, indicating the generic drug products are interchangeable for and substitutable with the RLD.
- BLA: Biological products are approved through the biologics license application (BLA). There is currently no provision analogous to section 505(b)(2) under the law governing the approval of biological products. While the FDA is authorized to approve biosimilars, including interchangeable biosimilars akin to generics, no current pathway permits the addition of new indications for use to a biosimilar product short of submitting a complete, original BLA with full preclinical and clinical data. Our proposed regulatory pathway is relevant for small molecule drugs and would not be applicable to biological products under current law.
Manufacturers can add new indications to their approved labeling without modifying the drug product through existing pathways. With supportive clinical evidence for the new indication, the NDA holder can file a supplemental NDA (sNDA), while an ANDA holder may submit a 505(b)(2) NDA as a supplement to their existing ANDA. As previously discussed, the drug product will likely be subject to pharmacy-level substitution with any available therapeutically equivalent generic. The marketing exclusivities that sponsors may receive from the FDA do not protect against this substitution. Therefore, these pathways are rarely, if ever, used by pharmaceutical companies when there are already multiple generic manufacturers of the product.
Challenges for non-manufacturers in using existing pathways
Since manufacturers are not incentivized to seek regulatory approval for new indications, labeling changes are more likely to happen if driven by non-manufacturers. Yet non-manufacturers face significant challenges in utilizing the existing regulatory pathways. Sponsors must submit the following information for all NDAs for the FDA’s review: 1) clinical and nonclinical data on the safety and effectiveness of the drug for the proposed indication; 2) the proposed labeling; and 3) chemistry, manufacturing, and controls (CMC) data describing the methods of manufacturing and the controls to maintain the drug product’s quality.
To submit and maintain NDAs, non-manufacturer sponsors would need to address the following challenges:
- Providing the FDA with required CMC data. NDA sponsors must provide CMC data for FDA review. Non-manufacturers would not produce physical drug products, and therefore they would not have information on the manufacturing process.
- Providing the FDA with product samples. If requested, NDA sponsors must have the drug products and other samples (e.g., drug substances or reference standards) available to support the FDA review process and must make available for inspection the facilities where the drug substances and drug products are manufactured. Non-manufacturers would not have physical drug products to provide as samples, the capabilities to produce them, or access to the facilities where they are made.
- Conducting post-marketing surveillance. Post-marketing responsibilities to maintain an NDA include conducting annual safety reporting and maintaining a toll-free number for the public to call with questions or concerns. Non-manufacturers, such as small nonprofits, may not have the bandwidth or resources to meet these requirements.
Within the current statutory framework, a non-manufacturer could sponsor a 505(b)(2) NDA to obtain approval of a new indication by partnering with a current manufacturer of the drug – either an NDA or ANDA holder. The manufacturer would help meet the technical requirements of the 505(b)(2) application that the non-manufacturer could not fulfill independently. Through this partnership, the non-manufacturer would acquire the CMC data and physical drug product samples from the manufacturer and rely on the manufacturer’s facilities to fulfill FDA inspection and quality requirements.
Once approved, the 505(b)(2) NDA would create a new drug product with indication-specific labeling, even though the product would be identical to an existing product under a previous NDA or ANDA. The 505(b)(2) NDA would then be tied to the specific manufacturer due to the use of their CMC data, and that manufacturer would be responsible for producing and distributing the drug product for the new indication.
As a practical matter, this pathway is rarely attainable. Manufacturers of marketed drug products, particularly generic drug manufacturers, lack the incentives needed to partner with non-manufacturers. Manufacturers may not want to provide their CMC data or samples because it may prompt FDA inspection of their facilities, require an update to their CMC information, or open the door to product liability risks. The existing incentive structure strongly discourages generic drug manufacturers from expending any additional resources on researching new uses or making any changes to their product labeling that would deviate from the original RLD product.
Plan of Action
To modernize drug labeling and enhance clinical adoption of generic drugs, the FDA should implement a dedicated regulatory pathway for non-manufacturers to seek approval of new indications for repurposed generic drugs. Ultimately, such a pathway would enable drug repurposing and be a crucial step toward equitable healthcare access for Americans. We propose a potential solution – a “labeling-only” 505(b)(2) NDA – as an extension of the existing 505(b)(2) approval pathway.
Overview of the proposed labeling-only 505(b)(2) NDA pathway
The labeling-only 505(b)(2) NDA would enable non-manufacturers to reference CMC information from previous FDA determinations and, when necessary, provide the FDA with samples of commercially available drug products. Through this approach, the new indication would not be tied to a specific drug product made by one manufacturer. There is no inherent necessity for a new indication of a generic drug to be exclusively linked to a single manufacturer or drug product when the FDA has already approved multiple therapeutically equivalent generic drugs. Any of these interchangeable drug products would be considered equally safe and effective for the new indication, and patients could receive any of these drug products due to pharmacy-level substitution.
We describe non-manufacturer repurposing sponsors as entities that intend to submit or reference clinical data through a labeling-only 505(b)(2) NDA. This pathway is designed to expand the FDA-approved labeling of generic drugs for new indications, including those that may already be considered the standard of care. Non-manufacturers do not have the means to independently produce or distribute drug products. Instead, they intend to show that there is substantial evidence to support the new use through FDA approval, and then advocate for the indication in clinical practice. This evidence may be based on their research or research performed by other entities, including clinical trials and real-world data analyses.
The labeling-only 505(b)(2) NDA pathway helps address the three major challenges non-manufacturers face in pursuing regulatory approval. Through this pathway, non-manufacturers would be able to:
- Reference the FDA’s previous determinations on CMC data. Currently, a 505(b)(2) NDA can reference the FDA’s previous determinations of safety and effectiveness for an approved drug product. For eligible generic drugs, the labeling-only 505(b)(2) NDA would build on this practice by allowing non-manufacturer sponsors to reference the FDA’s previous determinations on any NDA or ANDA that the manufacturing process and CMC data are adequate to meet regulatory standards.
- Provide the FDA with product samples using commercially available drug product samples. Currently, it is up to the discretion of the FDA whether or not to request samples in the review of an application. With the labeling-only 505(b)(2) NDA, non-manufacturers would provide the FDA with samples of commercially available products from generic manufacturers. Given that the FDA would have already evaluated the products and their bioequivalence to the RLD during the previous reviews, it is not expected that the FDA would need to re-examine the product at the level of requesting samples, except potentially to examine the packaging and physical presentation of the product for compatibility with the new indication and conditions of use. The facilities where the drugs are made would remain available for inspection, under the same terms and conditions as the existing, approved marketing applications.
- Manage post-marketing responsibilities. Since most post-marketing surveillance and adverse event reporting are drug product-specific, these obligations would continue to be the responsibility of the manufacturer of the physical drug product dispensed. With the labeling-only 505(b)(2) NDA, the non-manufacturer would not have product-specific obligations because they are not putting a new product into the marketplace. However, we anticipate the non-manufacturer would be responsible for the repurposed indication on their labeling, including but not limited to post-marketing surveillance as well as indication-specific adverse event reporting and reasonable follow-up.
Under the labeling-only 505(b)(2) NDA, the non-manufacturer sponsor would not introduce a new physical drug product into the market. The new labeling created by the approval would not expressly be associated with one specific product. The non-manufacturer’s labeling would refer to the drug by its established generic name. In that way, the non-manufacturer sponsor’s approval and labeling could be applicable to all equivalent versions of the drug product and would be available for patients to receive from their pharmacy in the same way that generic drugs are typically dispensed at the pharmacy. That is, with the benefit of pharmacy-level substitution, patients could receive any available, therapeutically equivalent drug products from any current manufacturer.
Eligibility criteria
We envision the users of this pathway to be non-manufacturers that conduct drug repurposing research for the public benefit, including organizations like nonprofits and patient advocacy groups. The FDA should implement and enforce additional guardrails on eligibility to ensure that sponsors operate in good faith and cannot otherwise meet traditional NDA requirements. This process may include pre-submission meetings and reviews. The labeling-only 505(b)(2) NDA should be held to the FDA’s standard level of rigor and scrutiny of safety and effectiveness for the proposed indication during the review process.
The labeling-only 505(b)(2) would only be suitable for well-established, commercially available small molecule generic drugs, which can be identified as:
- Drugs with a U.S. Pharmacopeia and National Formulary (USP-NF) monograph. The USP-NF monograph system ensures the uniformity of available products on the market by setting a consensus minimum standard of identity, strength, quality, and purity among all marketed versions of a drug. It is expressly recognized in the Federal Food, Drug, and Cosmetics Act (FDCA). The USP-NF strives to have substance and product monographs for all FDA-approved drugs. USP-NF monographs for generics are commonly available because the drugs have been on the market for a long time and are typically produced by multiple manufacturers. Drug products in the U.S. market must conform to the standards in the USP-NF, when available, to avoid possible charges of adulteration and misbranding.
By statute and regulation, the FDA already allows for NDAs and ANDAs to reference the USP-NF to satisfy some CMC requirements, such as for specifications of the drug substance. As an illustration of the acceptance of the USP-NF, clinical trial protocols requiring the use of background therapy or supportive care, as well as trials testing medical devices requiring the use of a drug product, often will specify that any available version of the drug product meeting USP-NF standards can be used. We propose that products without USP-NF monographs, including certain newer drugs and drugs with especially complicated manufacturing processes that are not conducive to standardization, would not be eligible for the labeling-only 505(b)(2) pathway.
- Drugs with multiple A-rated, therapeutically equivalent products in the FDA Orange Book. The FDA does not regulate which specific products are dispensed or substituted for a given drug prescription. The listing of therapeutic equivalents in the Orange Book facilitates the seamless replacement of drug products from different manufacturers in clinical practice. Therapeutically equivalent drug products: i) have demonstrated bioequivalence to the RLD; ii) have the same strength, dosage form, and route of administration as the RLD; and iii) are labeled for the same conditions of use as the RLD. Therapeutic equivalents that meet these criteria are designated “A-rated” in the Orange Book. A-rated drug products are substitutable for any other version of that A-rated drug product, including the RLD itself.
Implementation
The labeling-only 505(b)(2) NDA pathway could be implemented through an FDA guidance document interpreting the current statute and regulations or through legislation that clarifies the FDA’s existing authority. Guidance documents contain the FDA’s interpretation of a given policy on regulatory issues. The FDA’s Center for Drug Evaluation and Research (CDER) could issue new guidance that allows for interpretation of the existing statute, thereby officially authorizing previous FDA determinations of acceptable CMC data to be referenceable for eligible generic drugs and adjusting drug sample requirements. Alternatively, the labeling-only 505(b)(2) could be enacted by Congress through a statutory change by incorporation into FDCA, which is up for reauthorization through the Prescription Drug User Fee Act (PDUFA) in 2027, or other congressional acts as appropriate. FDA guidance would be a faster pathway to adoption, while statutory authorization would offer additional safeguards for the continuance of the pathway long term.
The labeling-only 505(b)(2) NDA pathway could be funded through user fees, which are established by PDUFA for the cost to file and maintain NDAs. However, many nonprofit sponsors would not be able to afford the same user fees as for-profit pharmaceutical manufacturers. Relevant statutes will likely need to be updated to create a different fee schema for non-manufacturers who use the labeling-only 505(b)(2) pathway. In a similar spirit to reducing barriers to maintaining up-to-date labeling, the 2017 PDUFA update waived the fee for submitting an sNDA, which is how an existing RLD holder would update their labeling with new indication information.
Conclusion
Patients need new and affordable treatment options for diseases that have a devastating societal impact, and repurposing generic drugs can help address this need. Nonprofits and other non-manufacturers are driving these efforts forward due to a lack of interest from pharmaceutical companies. As momentum gains for generic drug repurposing, the U.S. regulatory system needs a pathway for non-manufacturers to seek FDA approval of new indications for existing generic drugs. Our proposed labeling-only 505(b)(2) NDA would eliminate undue administrative burden, enabling non-manufacturers to pursue FDA approval of new indications. It would allow the FDA to provide the public with the most up-to-date drug labeling, improving the ability of patients and physicians to make informed treatment decisions. This dedicated pathway would increase the availability of effective treatment options while reducing costs for the American healthcare system.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
The FDA does not have sufficient bandwidth or resources to meet the opportunity we have with repurposed generic drugs. To be the primary driver of labeling changes for repurposed generic drugs, the FDA would need to identify repurposing opportunities and also thoroughly compile and evaluate the safety and effectiveness data for the new indications. Project Renewal in FDA’s Oncology Center of Excellence is working with RLD holders to update the labeling of certain older oncology drugs where the outdated labeling does not reflect their current clinical use. The initial focus of Project Renewal is limited, and newly repurposed treatments are not included within its scope. For newly repurposed treatments, the FDA could evaluate the drugs and post to the Federal Register reports on their safety and effectiveness that could be referenced by manufacturers. However, this approach is burdensome as it would require a significant commitment of FDA resources. By introducing a motivated, third-party non-manufacturer as the primary driver for labeling changes, non-manufacturers can contribute expertise and resources to enable faster data evaluation for more drugs.
The pre-existing NDA sponsor could update their labeling to add the new indication through an sNDA that references the labeling-only 505(b)(2) NDA. ANDA holders would then be legally required to match their labeling to that of the RLD. The FDA should determine whether all current manufacturers would be required to update their labeling following approval of the new indication, and if so, the appropriate process.
Generic drugs play a vital role in the U.S. healthcare system by decreasing drug spending and increasing the accessibility of essential medicines. Generics account for 91% of prescriptions filled in the U.S. Expanding the market with generics for new indications could lead to short-term price increases above the inflation rate for off-patent branded and generic drugs in some unlikely circumstances. For example, if a new use for a generic drug substantially increases demand for the drug, there is a short-term risk that prices for the drug or potential substitutes could rise until manufacturers build more capacity to increase supply. To mitigate this risk, generic manufacturers could be notified about potential increases in demand so they can plan for increased production.
Generally, healthcare payers are not required to cover or reimburse for off-label uses of drugs. Unless a drug undergoes utilization management, payers cover most generic drugs for off-label uses because coverage is agnostic of indication. Clinical practice guidelines are highly influential in the widespread adoption of off-label treatments into the standard of care and are often referenced by payers making reimbursement decisions. In oncology, many off-label treatments are included in guidelines; only 62% of treatments in the NCCN are aligned with FDA-approved indications. For example, more than half of the NCCN recommendations for metastatic breast cancer are off-label treatments. Due to the breadth of off-label use, we anticipate payers would continue to cover repurposed generic drugs used off-label, even if there is a pathway available for non-manufacturers to pursue FDA approval.
We do not envision any form of exclusivity being granted for indications pursued via a labeling-only 505(b)(2) NDA. Given that the non-manufacturer sponsor would rely on existing products produced by multiple generic manufacturers, there is no new product to grant exclusivity. Even if some form of exclusivity were given to the non-manufacturer, it would be insufficient to guarantee the use of any particular drug product over another due to pharmacy-level substitution.
Slow Aging, Extend Healthy Life: New incentives to lower the late-life disease burden through the discovery, validation, and approval of biomarkers and surrogate endpoints
The world is aging. Today, some two thirds of the global population is dying from an age-related condition. Biological aging imposes significant socio-economic costs, increasing health expenses, reducing productivity, and straining social systems. Between 2010 and 2030, Medicare spending is projected to nearly double – to $1.2 trillion per year. Yet the costly diseases of aging can be therapeutically targeted before they become late-stage conditions like Alzheimer’s. Slowing aging could alleviate these burdens, reducing unpaid caregivers, medical costs, and mortality rates, while enhancing productivity. But a number of market failures and misaligned incentives stand in the way of extending the healthy lifespan of aging populations worldwide. New solutions are needed to target diseases before they are life-threatening or debilitating, moving from retroactive sick-care towards preventative healthcare.
The new administration should establish a comprehensive framework to incentivize the discovery, validation, and regulatory approval of biomarkers as surrogate endpoints to accelerate clinical trials and increase the availability of health-extending drugs. Reliable biomarkers or surrogate endpoints could meaningfully reduce clinical trial durations, and enable new classes of therapeutics for non-disease conditions (e.g., biological aging). An example is how LDL (a surrogate marker of heart health) helped enable the development of lipid-lowering drugs. The current lack of validated surrogate endpoints for major late-life conditions is a critical bottleneck in clinical research. Because companies do not capture the majority of the benefit from the (expensive) validation of biomarkers, the private sector under-invests in biomarker and surrogate endpoint validation. This leads to countless lives lost and to trillions of public dollars spent on age-related conditions that could be prevented by better-aligned incentives. It should be an R&D priority for the new administration to fund the collection and validation of biomarkers and surrogate endpoints, then gain regulatory approval for them. As we explain below, the existing FNIH Biomarkers Consortium does not fill this role.
Currently, companies are understandably hesitant to invest in validation without clear rewards or regulatory pathways. The proposed framework would encourage private companies and laboratories to contribute their biomarker data to a shared repository. This repository would expedite regulatory approval, moving away from the current product-by-product assessment that discourages data sharing and collaboration. Establishing a broader pathway within the FDA for standardized biomarker approval would allow validated biomarkers to be recognized for use across multiple products, reducing the existing incentives to safeguard data while increasing the supply of validated biomarkers and surrogate endpoints. Importantly, this would accelerate the development of drugs which holistically extend the healthspan of aging populations in the U.S. by preventing instead of treating late-stage conditions. (Statins similarly helped prevent millions of heart attacks.)
Key players such as the FDA, NIH, ARPA-H, and BARDA should collaborate to establish a streamlined pathway for the collection and validation of biomarkers and surrogate endpoints, allowing these to be recognized for use across multiple products. This initiative aligns with the administration’s priorities of accelerating medical innovation and improving public health with the potential to add trillions of dollars in economic value by making treatments and preventatives available sooner. This memo outlines a framework applicable to various diseases and conditions, using biological aging as a case study where the validation of predictive and responsive biomarkers may be vital for significant breakthroughs. Other critical areas include Alzheimer’s disease and amyotrophic lateral sclerosis (ALS), where the lack of validated surrogate endpoints significantly hinders the development of life-saving and life-improving therapies. By addressing these bottlenecks, we can unlock new avenues for medical advancements that will profoundly improve public health and mitigate the fast-growing, nearly trillion-dollar Medicare spend on late-life conditions.
Challenge and Opportunity
By 2029, the United States will spend roughly $3 trillion dollars yearly – half its federal budget – on adults aged 65 and older. A good portion of these funds will go towards Medicare-related expenses that could be prevented. Yet the process of bringing preventative drugs to market is lengthy, costly, and currently lacking in commercial incentives. Even for therapeutics that target late-stage diseases, drug development often takes 10+ years and cost estimates range between $300 million to $2.8 billion. This extensive duration and expense are due, in part, to the reliance on traditional clinical endpoints, which require long-term observation and longitudinal data collection. The burden of chronic diseases is growing, and better biomarkers and surrogate endpoints are needed to accelerate the development of therapeutics that prevent non-communicable diseases and age-related decline. Chronological age, for instance, is a commonly used but inadequate surrogate marker for biological age. This means that, to date, clinical trials on therapeutics designed to improve the biology of aging take decades to validate, rather than years. As a result, pharmaceutical companies find more short-term rewards in treating late-stage diseases, since developing drugs that reduce overall age-related decline requires longer and currently uncertain endpoints.
The validation of reliable biomarkers and surrogate endpoints offers a promising solution to this challenge. Biological measures often correlate with and predict clinical outcomes, and can therefore provide early indications of whether a treatment is effective. If sufficiently predictive, biomarkers can serve as surrogate clinical endpoints, potentially reducing the duration and cost of clinical trials. Validated biomarkers must accurately predict clinical outcomes and be accepted by regulatory authorities, yet the validation process is underfunded due to insufficient commercial incentives for individual agents to share their biomarkers to be used as a public good. (From a purely financial standpoint, companies are better off targeting diseases with known endpoints.)
The most prominent existing efforts to advance biomarkers and surrogate endpoints are the Foundation for the National Institute of Health’s (FNIH) Biomarkers Consortium and the FDA’s Biomarker Qualification Program. Established in 2006, the Biomarkers Consortium is a public-private partnership aimed at advancing the development and use of biomarkers in medical research. Meanwhile, the FDA’s qualification program was the result of the 21st Century Cures Act, passed in 2016, which underscored the critical role biomarkers play in accelerating medical product development. The Act mandated the FDA to implement a more transparent and efficient process for biomarker qualification.
Despite the Consortium’s ambitious goals, the rate of biomarker qualification by the FDA has been slow. Since its inception in 2006, only a small number of biomarkers have been successfully qualified. This sluggish progress has been a source of criticism for stakeholders, especially given the high level of resources and collaboration involved. For example, the process of validating biomarkers for osteoarthritis under the Consortium’s “PROGRESS OA” project has been ongoing since Phase 1 and still faces hurdles before full qualification. We are of the view that this is the result of two issues. Firstly, the qualification process, which involves FDA approval, is seen as overly complex and time-consuming. Despite the 21st Century Cures Act aiming to streamline the process, resulting in the qualification pathway, it remains a significant challenge. The difficulty in navigating the regulatory landscape can limit the impact of Biomarkers Consortium (BC) projects. The Kidney Safety Project, for example, faced substantial regulatory hurdles before finally achieving the first qualification of a clinical safety biomarker. Secondly, even though the Consortium operates in a precompetitive space, there are ongoing challenges related to data sharing. Companies may still hesitate to share critical data that could advance biomarker validation out of concern for losing a competitive edge, which hampers collaboration. To address these issues, it is crucial to implement a framework that promotes data sharing in the academic and private sectors, providing strong incentives for the validation and regulatory approval of biomarkers, while improving regulatory certainty with a standardized regulatory process for surrogate endpoint validation.
The current boom in biotechnology underscores the urgency of addressing persisting inefficiencies. Without changes, we face a significant bottleneck in proving the efficacy of new drugs. This is exacerbated by Eroom’s Law—the observation that drug discovery is becoming slower and more expensive over time. This growing inefficiency threatens to hinder the development of new, life-saving treatments at a time when the American population is aging and rapid medical advancements are crucial to deter increasing medical and social costs. In just 11 years—between 2018 and 2029—the U.S. mandatory spending on Social Security and Medicare will more than double, from $1.3 trillion to $2.7 trillion per year. Yet the costly diseases of aging can be therapeutically targeted before they become late-stage conditions like Alzheimer’s. For federal policymakers, taking immediate action to improve data sharing and biomarker validation processes is vital. Failure to do so will not only stifle innovation but also delay the availability of critical therapies that could save countless lives and accelerate economic growth in the long run. Prompt policy intervention is essential to capitalize on the current advancements in biotechnology and ensure the development of new life-saving tests, tools, and drugs.
Implementing pull-incentives for data sharing now can help the United States adjust to its new demographic structure, where adults in advanced age prevail, while fertility rates decline. It can also mitigate the escalating costs and timelines of clinical trials, and accelerate the approval of life-saving, health-extending drugs. If our proposed framework is successfully implemented, a robust pool of biomarker data will be established, significantly facilitating the discovery and validation of biomarkers. This will result in several key advancements, including shortened clinical trial durations, increased R&D investment, faster drug approvals, and even increased drug efficacy. Additionally, new drug classes targeting non-disease endpoints, such as biological aging, could be developed. Just as the discovery of LDL as a surrogate marker of heart health was critical in enabling the testing and development of statins, the discovery of clinical-grade biomarkers may unlock new therapeutics designed to target the mechanisms that drive human aging, slowing down the progression of age-related diseases (like cancers) before they become deadly and socio-economically expensive.
Plan of Action
To address the challenge of inefficient data sharing, validation, and approval of biomarkers, we propose implementing a series of pull-incentives aimed at encouraging pharmaceutical companies to contribute their relevant biomarker data to a shared repository and undertake the necessary research and analysis for public validation. These validated biomarkers can then be formally accepted by regulators as surrogate endpoints for drug approval, accelerating the drug development process and reducing late-life costs.
Recommendation 1. An NIH-FDA initiative for Biomarkers and Surrogate Endpoints Within the NIA
Most existing agencies focus on single, often late-stage diseases. This is at odds with a holistic understanding of human biology. A new initiative within the National Institute on Aging (NIA) could be devoted to the discovery, collection, and validation of biomarkers and surrogate endpoints for overall human health and age-related decline. Most National Institutes of Health funds are currently devoted to the diseases of aging (think cancers, Alzheimer’s, heart disease, or Parkinson’s.) Within the NIA, research on Alzheimer’s disease alone receives roughly eight times more funding than the biology of aging, with few human-relevant results. Every federal agency and U.S. individual would benefit from better biomarkers of long-term health and from an understanding of how to measure the biology of aging. Yet no single agent has the incentives to collect and validate this data, for instance by shouldering the costs of validating predictive and responsive biomarkers of aging.
This new initiative could also be devoted to the development of preclinical, human-relevant methodologies that could broadly facilitate or streamline drug development. In 2022, the FDA Modernization Act 2.0 approved the use of in vitro and in silico New Approach Methodologies (NAMs) like cell-based assays (e.g. organs-on-chips) or computer models (like virtual cells) in preclinical development to reduce or replace animal studies, especially “where no pharmacologically relevant animal species exists.” This may be the case for human aging, where no single animal model reflects the full complex biology of our aging process.
At present, these technologies cannot accurately represent the multifactorial processes of aging, and they cannot model entire organisms. Much work remains to be done to even understand how to “code” aging into organs-on-chips. Yet if supplemented by approaches like in vivo pooled screening, next generations of human-relevant in vitro or in silico methodologies (like virtual cells) could be infused with the complex data needed to accelerate clinical trial results and increase drug efficacy. For in vitro and in silico models to reproduce key aspects of aging biology, a better understanding of how human aging works in living organisms — and what markers to include to represent it either virtually or in vitro — may be needed. Yet pharmaceutical companies, startups, health insurance firms, and even research hospitals again lack the incentives to shoulder the costs of collecting and validating this type of data. This means a new office within a federal agency may be needed to supply these incentives.
Recommendation 2. New Data-sharing Incentives
The specific incentives used would need to be developed in collaboration with policymakers and industry stakeholders, but a few are outlined below:
Pull-incentives
One possibility is offering transferable Priority Review Vouchers (PRVs) or similar pull incentives to companies that share their biomarker data. PRVs are currently awarded by the FDA to companies developing drugs for neglected tropical diseases, rare pediatric diseases, or medical countermeasures. A PRV allows the holder to expedite the FDA review of another drug from 10 months to 6 months, and holds significant financial value. Offering transferable PRVs for drugs designed to target biological aging, for instance, could create the incentives needed for pharmaceutical companies to target early-stage age-related conditions before they turn into diseases.
The creation of a new PRV category would require legislative action. Our proposed NIH-FDA initiative would be well positioned to oversee the issuance of PRVs, working with government agencies and think tanks to determine, for instance, what an “aging therapeutic” means, and what a company needs to achieve to gain a PRV for a longevity drug. The Alliance for Longevity Initiatives, for instance, has developed an advanced approval pathway for health-extending drugs that directly target the biology of aging. Another possible strategy would be for the FDA to encourage drugs that target multiple disease indications at once, perhaps offering discounts or incentives for every extra biomarker or surrogate endpoint validated. This could effectively encourage the development of drugs that do more than marginally improve on existing interventions.
We acknowledge that an overabundance of PRVs can saturate the market, decreasing their value and weakening the intended pull-incentive for pharmaceutical innovation. A response would be to demand that proposals to issue additional PRVs include a comprehensive market impact analysis to mitigate unintended economic consequences. Expanding the number of PRVs can also place extra demands on the FDA’s limited resources, potentially leading to longer approval times for other essential medications, even though PRV holders often delay redemption, preventing an immediate influx of priority review applications. The PRV system may inadvertently favor larger, well-established pharmaceutical companies that have the means to acquire and leverage PRVs effectively, creating barriers for smaller firms and startups. These are all spill-over problems worth solving for the potential upshot of mitigating late-life disease costs and encouraging drugs that holistically improve the human healthspan.
Biomarker Data Sharing as a Condition of Federal Funding
Federal funding recipients are legally obligated to make their research publicly accessible through agency-specific policies aimed at advancing open science. This mandate was strengthened by the 2022 OSTP Memorandum. Despite this clear mandate, the implementation of public access policies has been uneven across federal agencies, with progress varying due to differences in resources, technical infrastructure, and agency-specific priorities. The 2022 OSTP Public Access Memorandum aims to accelerate agency efforts to enhance public access infrastructure and policies. This updated guidance presents an opportunity for agencies to not only meet immediate data-sharing requirements but also to expand policy scopes to include essential clinical data, such as biomarker data from clinical trials. To meet these goals, agencies should ensure that funding agreements explicitly require the publication of comprehensive biomarker data and that suitable repositories are available to store and share these critical datasets effectively.
Case Study: Project NextGen
A prime example of the potential success of such initiatives is Project NextGen, a program led by BARDA in collaboration with the NIH to advance the next generation of COVID-19 vaccines and treatments. As part of its vaccine program, Project NextGen includes centralized immunogenicity assays with the overarching goal of establishing correlates of protection, which could serve as surrogate biomarkers for next-gen vaccines. These assays are collected during Phase 2b vaccine studies sponsored by Project NextGen, which have been designed to measure a number of secondary immunogenicity endpoints including systemic and mucosal immune responses. Developers share their assays so that they can be used as a public good, in return for federal funding. This effort demonstrates the feasibility and benefits of a federally led effort to share assay data to advance biomarker validation and drug development.
Recommendation 3. Create and Manage a Data Repository
To enhance collaborative research and ensure the efficient use of publicly funded clinical data, we recommend establishing a secure data repository. This repository will serve as a centralized platform for data submission, storage, and access. Management of the repository could be undertaken by a federal agency, such as the NIH, leveraging their experience with the Biomarkers Consortium, perhaps in partnership with non-governmental organizations like the Biomarkers of Aging Consortium. Drawing from existing models, such as Project NextGen’s assay data management, can provide valuable insights into the implementation and operationalization of the repository.
The cost of establishing and maintaining this repository, including data storage, management, and access controls, would be dwarfed by the socio-economic returns it could provide. This repository can facilitate data sharing, protect sensitive information, and promote a collaborative environment that accelerates biomarker validation and approval, while ensuring pharmaceutical companies that their hard-earned data is safely stored.
The securely stored data in the repository would primarily be accessible to qualified researchers, clinicians, and policymakers involved in biomarker research and development, including academic researchers, pharmaceutical companies, and public health agencies. Access would be granted through an application and review process. The benefits of this repository are multifaceted: it accelerates research by providing a centralized database, enhances collaboration among scientists and institutions, increases efficiency by reducing redundancy and improving data management, ensures data security through robust access controls, offers cost-effectiveness with long-term socio-economic returns, and supports regulatory bodies with comprehensive data sets for more informed decision-making.
Recommendation 4. Create A Regulatory Pathway with Broader Application
To accelerate the adoption of validated biomarkers and surrogate endpoints in drug development, we propose the creation of a streamlined regulatory approval process within the FDA. This new pathway would establish clear criteria and standardized procedures for biomarker evaluation and approval, facilitating their recognition for use across multiple products and therapeutic areas.
Currently, the FDA’s Center for Drug Evaluation and Research (CDER) operates the Biomarker Qualification Program (BQP), which allows drug developers to seek regulatory qualification for specific contexts of use. While this program fosters collaboration between the FDA and external stakeholders, biomarkers are qualified on a case-by-case basis, limiting their broader applicability across different drug development programs.
Additionally, the FDA maintains a Table of Surrogate Endpoints that have been used as the basis for drug approvals under the accelerated approval pathway. However, this table primarily serves as a reference and does not comprehensively address the need for a streamlined approval process for biomarkers and surrogate endpoints.
By developing a framework that moves away from traditional product-by-product assessments, the FDA could reduce existing barriers to biomarker and surrogate endpoint discovery and approval. This approach would encourage data sharing and collaboration among pharmaceutical companies and research institutions, leading to faster validation and broader acceptance of these critical tools in drug development.
This proposal builds upon existing legislative efforts, such as the 21st Century Cures Act of 2016, which includes provisions to accelerate medical product development and supports the use of biomarkers and surrogate endpoints in the regulatory process. Furthermore, it aligns with the FDA’s ongoing efforts to provide clarity on evidentiary criteria for biomarker qualification, as outlined in the 2018 guidance document “Biomarker Qualification: Evidentiary Framework.”
Inspiration for this approach can be drawn from the Advanced Approval Pathway for Longevity Medicines (AAPLM) proposed by the Alliance for Longevity Initiatives (See AAPLM-Whitepaper). The AAPLM includes provisions such as a special approval track, a priority review voucher system, and indication-by-indication patent term extensions, which align economic incentives with the transformative health improvements that longevity medicines can provide. These measures offer a valuable template for facilitating the recognition and approval of biomarkers. Adding to the existing FDA table of surrogate endpoints that can serve as the basis for drug approval or licensure, and referencing existing collaborations between the NIH and FDA, such as the Biomarkers Consortium, can provide a robust foundation for new biomarker evaluations. Ultimately, this regulatory innovation will support the development of life-saving drugs, enhance public health outcomes, and meaningfully contribute to economic growth by bringing effective treatments to market more quickly.
Conclusion
Today, over two thirds of all deaths in the United States are the result of an age-related condition. The burden of non-communicable diseases is growing, and better biomarkers and surrogate endpoints are needed to target diseases before they are life-threatening or debilitating. The next administration should implement a comprehensive framework to promote data sharing and incentivize the validation and regulatory approval of biomarkers and surrogate endpoints. This aligns directly with the administration’s goal to make Americans healthy. These solutions can substantially reduce the duration and cost of clinical trials, accelerate the development of life-saving drugs, and improve public health outcomes. It is possible and necessary to create an environment that encourages and rewards pharmaceutical companies to share crucial data that accelerates medical innovation. By discovering and validating predictive and responsive biomarkers of health and disease, new therapeutic classes can be developed to directly target biological aging and prevent most forms of cancers, heart disease, frailty, vulnerability to severe infection, and Alzheimer’s. This will enable the United States to remain at the forefront of medical research, and to respond to the growing demographic crisis of aging populations in declining health.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
A number of market failures stand in the way of the discovery and validation of predictive, reliable, and responsive biomarkers. First, it’s currently expensive to test drugs in multiple disease indications, which means pharmaceutical companies are often incentivized to focus on late-stage diseases (e.g. delaying death by a terminal cancer by three months), since this drug class is more easily and quickly trialed. The FDA also strongly assumes that a treatment ought to modulate a single outcome. (Think life/death; heart disease/no heart disease.) Therapeutics that target biological aging, for instance, would take decades to test without validated biomarkers or widely accepted surrogate endpoints.
Aging research, for instance, has seen a 70-fold increase in venture capital funding since the last decade. Yet so far—and this is a critical asterisk—misaligned commercial incentives have mostly optimized for unproven supplements, imprecise biological-age-tracking apps, and unsafe experimental therapies or cosmetics. The most well-meaning investors and founders in “longevity” often end up developing drugs for single disease indications (like osteoarthritis, or obesity) to avoid bankruptcy or as a path to self-fund their intent of developing drugs that more holistically target the mechanisms that drive aging. arket incentives need to be aligned to the pressing social needs these therapeutics could respond to.
The federal government is uniquely positioned to coordinate large-scale initiatives that require significant resources and regulatory oversight. While private sector companies play crucial roles in drug development, they often lack the incentives to self-coordinate and the authority to drive comprehensive data-sharing and biomarker validation efforts.
Cohesion from data-collection to regulatory approval of biomarkers is going to be key if surrogate endpoints are actually going to be adopted. Having the federal government oversee all stages will ensure this cohesion.
The Biomarkers Consortium has made meaningful strides in advancing biomarker research, but they have not succeeded in acquiring sufficient data. The consortium relies on voluntary, precompetitive collaboration without providing strong financial or legislative incentives for data sharing. It does not maintain a centralized, secure data repository, and struggles with fragmented data sharing. It also lacks influence over the FDA’s biomarker qualification process, which remains complex and time-consuming. This has resulted in slow progress due to hesitancy from private entities to share valuable data. Our solution differs by directly addressing this data-sharing hurdle through a series of targeted incentives that reduce the case-by-case assessment currently required, and enable broader application of validated biomarkers across multiple drugs and therapeutic areas.
By introducing legislative changes to authorize patent extensions and expand Priority Review Vouchers (PRVs), we create compelling reasons for companies to share their data. Additionally, our proposal includes the development of a centralized data repository with a streamlined regulatory approval process, inspired by the Advanced Approval Pathway for Longevity Medicines (AAPLM). This approach not only incentivizes data sharing but also provides a clear and efficient pathway for biomarker validation and regulatory acceptance. By leveraging existing frameworks and offering tangible rewards, our solution proposes an increase in incentives, to match the socioeconomic benefits that may be unlocked by more accessibility to the wealth of existing but undersupplied biomedical data.
The FDA’s Accelerated Approval Pathway is indeed a valuable tool that allows for the approval of drugs based on surrogate endpoints that are reasonably likely to predict clinical benefit. This pathway requires substantial evidence showing that these surrogate endpoints are linked to clinical outcomes, usually gathered from rigorous clinical trials. However, it typically applies to surrogate endpoints validated for specific uses or products. Our goal is to establish a new pathway that supports the validation and use of surrogate endpoints across multiple products. By validating biomarkers that can be used across various drugs, we can streamline the drug development process, reducing the time and cost associated with bringing new therapies to market. This broader approach would enhance efficiency, reduce drug development time and costs, and promote innovation by encouraging pharmaceutical companies to invest in research, knowing that successful biomarkers can have wide-reaching applications.
Pharmaceutical companies could push back on this proposal due to concerns over losing their competitive advantage by sharing proprietary data. They might reasonably fear that sharing valuable biomarker data could erode their market position and intellectual property. By involving pharmaceutical companies in the development of the proposal, we can better understand their concerns and tailor incentives accordingly. One effective strategy would be to offer significant financial incentives, such as Priority Review Vouchers (PRVs) or patent term extensions to companies that share their data. These incentives can offset the perceived risks and provide tangible benefits that make data sharing more attractive. By making PRVs transferable and offering additional incentives to small biotechnology companies, this policy can be implemented without overly favoring large pharmaceutical companies. Another possible strategy would be for the FDA to encourage drugs that target multiple disease indications at once, perhaps offering discounts or incentives for every extra biomarker or surrogate endpoint validated. Fostering a collaborative environment where the benefits of shared data (such as accelerated drug approvals and reduced R&D costs) are clearly communicated can reduce hurdles. Engaging economists to quantify the long-term economic gains to individual pharmaceutical companies as well as to society, while demonstrating how shared data can lead to industry-wide advancements, can further encourage participation. By providing competitive enough incentives, a framework can be created that balances the interests of pharmaceutical companies with the broader goal of advancing medical innovation and public health.
The first step to get this proposal off the ground is to introduce legislative changes that authorize patent extensions and expand the eligibility for Priority Review Vouchers (PRVs). These legislative changes will create the necessary incentives for pharmaceutical companies to participate in the program by offering tangible benefits that offset the risks associated with data sharing.
Simultaneously, developing and launching a pilot program for the centralized data repository is crucial. This pilot should focus on a specific subset of biomarkers for high-priority diseases and non-disease indications to demonstrate the feasibility and benefits of the proposed framework. By starting with a targeted approach, we can gather initial data, test the processes, and make any necessary adjustments before scaling up the program. This pilot will not only help in garnering support from stakeholders by showcasing the practical benefits of the framework but also refine the approach based on real-world feedback, ensuring a smoother and more effective broader implementation.
Similar efforts in the past have often been hindered by a lack of incentives for data sharing and collaboration, along with fragmented regulatory processes. Our proposal aims to overcome these obstacles by introducing strong incentives which will encourage companies to share their data. Moreover, we propose creating a standardized regulatory pathway for biomarker approval, which will streamline the process and reduce fragmentation. By involving key federal agencies, we ensure a coordinated and comprehensive implementation, thus avoiding the pitfalls that have doomed past efforts.
The status quo is unacceptable. Millions of lives are lost or debilitated every year due to the slow and costly process of bringing new drugs to market, which is hindered by the lack of validated biomarkers and surrogate endpoints. The recommended course of action leverages existing regulatory frameworks and incentives that have proven effective in other contexts, such as the use of Priority Review Vouchers (PRVs) for neglected tropical diseases. By adapting these mechanisms to encourage data sharing and biomarker validation, we can build on established successes while addressing the specific challenges of the current drug development landscape.
This approach ensures that we utilize proven strategies to accelerate drug development and approval, reducing the overall time and cost associated with clinical trials. By fostering a collaborative environment and providing tangible incentives, we can significantly enhance the efficiency and effectiveness of the drug development process. This targeted strategy not only addresses the immediate needs but also sets a foundation for continuous improvement and innovation in the field of medical research, ultimately saving lives and improving public health outcomes.
The Medicare Advance Healthcare Directive Enrollment (MAHDE) Initiative: Supporting Advance Care Planning for Older Medicare Beneficiaries
Taking time to plan and document a loved one’s preferences for medical treatment and end-of-life care helps respect and communicate their wishes to doctors while reducing unnecessary costs and anxiety. There is currently no federal policy requiring anyone, including Medicare beneficiaries, to complete an Advance Healthcare Directive (AHCD), which documents an individual’s preferences for medical treatment and end-of-life care. At least 40% of Medicare beneficiaries do not have a documented AHCD. In the absence of one, medical professionals may perform major and costly interventions unknowingly against a patient’s wishes.
To address this gap, the Centers for Medicare and Medicaid Services (CMS) should launch the Medicare Advance Healthcare Directive Enrollment (MAHDE) Initiative to support all adults over age 65 who are enrolled in Medicare or Medicare Advantage plans to complete and annually renew, at no extra cost, an electronic AHCD made available and stored on Medicare.gov or an alternative secure digital platform. MAHDE would streamline the process and make it easier for Medicare enrollees to complete and store directives and for healthcare providers to access them when needed. CMS could also work with the National Committee for Quality Assurance (NCQA) to expand Advance Care Planning (ACP) Healthcare Effectiveness Data and Information Set (HEDIS) measures to include all Medicare Advantage plans caring for beneficiaries aged 65 and older.
AHCDs save families unnecessary heartache and confusion at times of great pain and vulnerability. They also aim to improve healthcare decision-making, patient autonomy, and function as a long-term cost-saving strategy by limiting undesired medical interventions among older adults.
Challenge and Opportunity
Advance healthcare directives document an individual’s preferences for medical treatment in medical emergencies or at the end of life.
AHCDs typically include two parts:
- Identifying a healthcare proxy or durable power of attorney, who will make decisions about an individual’s health when they are unable to.
- A living will, which describes the treatments an individual wants to receive in emergencies—such as CPR, breathing machines, and dialysis—as well as decisions on organ and tissue donation.
Other documents complement AHCDs and help communicate treatment wishes during emergencies or at the end of life. These include do-not-resuscitate orders, do-not-hospitalize orders, and Physician (or Medical) Orders for Life-Sustaining Treatment forms, along with similar portable medical order forms for seriously ill or frail individuals (e.g., Medical Orders for Scope of Treatment, Physician Orders for Scope of Treatment, and Transportable Physician Orders for Patient Preferences). These forms are all designed to honor an individual’s healthcare preferences in a future medical emergency.
With the U.S. aging population projected to reach more than 20% of the total population by 2030, addressing end-of-life care challenges is increasingly urgent. As people age, their healthcare needs become more complex and expensive. Notably, 25% of all Medicare spending goes toward treating people in the last 12 months of their life. However, despite commonly receiving more aggressive treatments, many older adults prefer less intensive medical interventions and prioritize quality of life over prolonging life. This discrepancy between care received and a patient’s wishes is common, highlighting the need for clear and proactive communication and planning around medical preferences. Research shows patients with ACPs are less likely to receive unwanted and aggressive treatments in their last weeks of life, are more likely to enroll in hospice for comfort-focused care, and are less likely to die in hospitals or intensive care units.
Established ACP Policies and Support Mechanisms
Historically, some federal policies have underscored the importance of patient decision-making rights and the role of AHCDs in helping patients receive their desired care. These policies reflect the ongoing effort to empower patients to make informed decisions about their healthcare, particularly in end-of-life situations.
The Patient Self-Determination Act (PSDA), a federal law introduced in 1990 as a part of the Omnibus Budget Reconciliation Act, was created to ensure that patients are informed of their rights regarding medical care and their ability to make decisions about that care, especially in situations where they are no longer able to make decisions for themselves.
The Act requires hospitals, skilled nursing facilities (SNFs), home health agencies, hospice programs, and health maintenance organizations to:
- Inform patients of their rights to make decisions under state law about their medical care, including accepting or refusing treatment.
- Periodically inquire whether a patient has completed a legally valid AHCD and make note in their medical record.
- Not discriminate against patients who do or do not have an advance directive.
- Ensure AHCDs and other documented care wishes are carried out, as permitted by state law.
- Provide education to staff, patients, and the community about AHCDs and the right to make their own medical decisions.
It also directs the Secretary of Health and Human Services to research and assess the implementation of this law and its impact on health decision-making. Additionally, to encourage physicians and qualified health professionals to facilitate ACP conversations and complete AHCDs, CMS introduced and approved two new billing codes in 2016, allowing qualified health providers to bill CMS for advance care planning as a separate service regardless of diagnosis, place of service, or how often services are needed (Figure 1). These codes were expanded in 2017 with the temporary Healthcare Common Procedure Coding System code G0505, followed by CPT code 99483, to offer care planning and cognitive assessment services that include advance care planning for Medicare beneficiaries with cognitive impairment.

Figure 1. Two primary CPT codes and billing descriptors for advance care planning reimbursement. (Source: CMS Medicare Learning Network Fact Sheet)
In 2022, ACP was introduced as one of four key components of the Care for Older Adults (COA) initiative within the Healthcare Effectiveness Data and Information Set measures. HEDIS is a proprietary set of clinical care performance measures developed by the National Committee for Quality Assurance (NCQA), a private, nonprofit accreditation organization that creates standardized measures to help health plans assess and report on the quality of care and services. HEDIS evaluates areas such as chronic disease management, preventive care, and care utilization, enabling reliable comparisons of health plan performance and identifying areas for improvement. It is reported that 235 million Americans are enrolled in plans that report HEDIS results, and reporting HEDIS measures is mandatory for Medicare Advantage plans.
The COA initiative includes the ACP measure as a reporting requirement for Medicare Special Needs Plans (SNPs), which are plans designed for individuals who have complex care needs, are eligible for Medicare and Medicaid, have disabling or chronic conditions, and/or live in an institution. The report includes the percentage of Medicare Advantage members within a specified population who participate in ACP discussions each year. This population currently includes:
- Adults aged 65–80 with advanced illness, signs of frailty, or those receiving palliative care.
- All adults aged 81 and older.
HEDIS measures contribute to the overall STAR rating of Medicare Advantage and other health plans, which helps beneficiaries choose high-quality plans, enables highly rated plans to attract more members, and influences the funding and bonuses CMS provides to these plans.
Despite the PSDA, CMS provider reimbursement codes that incentivize physicians and qualified health professionals to facilitate advance care planning, and its recent inclusion in HEDIS measures for Medicare Special Needs Plans, there remain many barriers to completing AHCDs.
Barriers to AHCD Completion
Although Medicare provides health and financial security to nearly all Americans aged 65 and older, completing a comprehensive AHCD is not universally expected within this population. Conversations about treatment decisions in future emergencies and end-of-life care are often avoided for various cultural, religious, financial, and mental health reasons. When they do happen, preferences are more often shared with loved ones but not documented or communicated to healthcare professionals. It is perhaps unsurprising, then, that only about half of Medicare beneficiaries have completed an AHCD. Studies show that of those who have, most do so in conjunction with estate planning, which may explain increasing cultural and socioeconomic disparities in the completion of AHCDs.
For Medicare beneficiaries who wish to complete an AHCD with a physician or qualified health professional, Medicare only covers planning as part of the annual wellness visit. If ACP is provided outside of this visit, the beneficiary must meet the Medicare Part B deductible, which is $240 in 2024, before coverage begins. If the deductible has not been met through other Part B services (such as doctor visits, preventive care, mental health services, or outpatient procedures), the beneficiary is responsible for the deductible and a 20% coinsurance payment. Additionally, some states may require attorney services or notarization to legally validate an AHCD, which could incur extra costs.
These additional costs can make it challenging for many Medicare beneficiaries to complete an AHCD when they want to. Furthermore, depending on the complexity of their situation and readiness to make decisions, many patients may need more than one visit with their clinical provider to make decisions about critical illness and end of life care, creating more out-of-pocket expenses.
AHCDs can also vary widely, are not uniform across states, and are often stored in paper formats that can be easily lost or damaged, or are embedded in bulky, multipage estate plans. Efforts to centralize AHCDs have been made through state-based AHCD registries, but their availability and management vary significantly and there is limited data on their use and effectiveness. Additionally, private ACP programs through initiatives like the Uniform Health-Care Decisions Act (UHCDA), the Five Wishes program, MyDirectives, and the U.S. Advance Care Plan Registry (USACPR), among others, have contributed to broadening ACP accessibility and awareness. However, data from private ACP programs are not widely published or have shown variable results. The UHCDA, first drafted and approved in 1993 by the Uniform Law Commission—a nonprofit organization focused on promoting consistency in state laws—was updated in 2023 and aims to address these variations in state AHCD policies, however with varying degrees of success. The Five Wishes program reports 40 million copies of their paper and digital advance directives in circulation nationwide, and their digital program recently launched a partnership with MyDirectives, a leader in digital advance care planning, to facilitate electronic access to legally recognized ACP documents. Unfortunately, data on completion and storage of these directives is not consistently reported across all users.
Despite efforts by numerous organizations to improve ACP completion, access, and usability, the lack of updated federal policy supporting advance care planning makes it difficult for patients to complete them and healthcare providers to quickly locate and interpret them in critical situations. When AHCDs are not available, incomplete, or hard to find, medical professionals may be unaware of patients’ care preferences during urgent moments, leading to treatment decisions that may not align with the patients’ wishes.
Plan of Action
To support all Medicare beneficiaries aged 65 and older in documenting their end-of-life care preferences, encourage the completion of AHCDs, and improve accessibility of AHCDs for healthcare professionals, CMS should launch the Medicare Advance Healthcare Directive Enrollment Initiative to focus on the following four interventions.
Recommendation 1. Streamline the process of AHCD completion and electronic storage during open enrollment through Medicare.gov or an alternative CMS-approved secure ACP digital platform.
To provide more clarity and support to fulfill patients’ wishes in their end-of-life care, CMS should empower all adults over the age of 65 enrolled in Medicare and Medicare Advantage plans to complete an electronic AHCD and renew it annually, at no extra cost, during Medicare’s designated open enrollment period. Though electronic completion is preferred, paper options will continue to be available and can be submitted for electronic upload and storage.
Supporting the completion of an AHCD during open enrollment presents a strategic opportunity to integrate AHCD completion into overall discussions about healthcare options. New open enrollment tools can be made easily available on Medicare.gov or in partnership with an existing digital ACP platform such as the USACPR, the newly established Five Wishes and MyDirectives partnership, or a centralized repository of state registries, enabling beneficiaries to complete and safely store their directives electronically. User-friendly tools and resources should be tailored to guide beneficiaries through the process and should be age-appropriate and culturally sensitive.
Building on this approach, some states are also taking steps to integrate electronic ACP completion and storage into healthcare enrollment processes. For example, in 2022, Maryland unanimously passed legislation that mandates payers to offer ACP options to all members during open enrollment and at regular intervals thereafter. It also requires payers to receive notifications on the completion and updates of ACP documents. Additionally, providers are required to use an electronic platform to create, upload, and store AHCDs.
An annual electronic renewal process during open enrollment would allow Medicare beneficiaries to review their own selections and make appropriate changes to ensure their choices are up to date. The annual review will also allow for educational opportunities around the risks and benefits of life-extending efforts through the secure Medicare enrollment portal and is a time interval that accounts for the abrupt changes in health status as individuals age. The electronic enhancements also provide a better fit with the modern technological healthcare landscape and can be completed in person or via a telehealth ACP visit with a physician or qualified health professional. Updates to AHCDs can also be made at any time outside of the open enrollment period.
CMS could also work across state lines and in collaboration with private ACP organizations, the UHCDA, and state-appointed AHCD representatives to develop a universal template for advance directives that would be acceptable nationwide. Alternatively, Medicare.gov could provide tailored, state-specific electronic forms to meet state legal requirements, like the downloadable forms provided by organizations such as AARP, a nonprofit, nonpartisan organization for Americans over 50, and CaringInfo, a program of the National Hospice and Palliative Care Organization. Either approach would ensure AHCDs are legally compliant while centralizing access to the correct forms for easy completion and secure electronic storage.
Recommendation 2. Remove barriers to access advance care planning services.
CMS should remove the deductible and 20% coinsurance when beneficiaries engage in voluntary ACP services with a physician or other qualified health professional outside of their yearly wellness visit.
The current deductible and coinsurance requirements may discourage participants from completing their AHCDs with the guidance of a medical provider, as these costs can be prohibitive. This is similar to how higher cost-sharing and out-of-pocket health expenses often result in cost-related nonadherence, reducing healthcare engagement and prescription medication adherence. When individuals face higher out-of-pocket costs for care, they are more likely to delay treatments, avoid doctor visits, and fill fewer prescriptions, even if they have insurance coverage. Removing deductibles and coinsurance for ACP visits would allow individuals to complete or update their AHCDs as needed, without financial strain and with support from their clinical team, like preventive services.
Additionally, CMS could consider continued health provider education on facilitating ACPs and partnership with organizations like Institute of Healthcare Improvement’s The Conversation Project, which encourages open discussions about end-of-life care preferences. Partnering with Evolent (formerly Vital Decisions) could also support ongoing telehealth discussions between behavioral health specialists and older adults, focusing on late-life and end-of-life care preferences to encourage formal AHCD completion. Internal studies of the Evolent program, aimed at Medicare Advantage beneficiaries, demonstrated an average savings of $13,956 in the final six months of life and projected a potential Medicare spending reduction of up to $8.3 billion.
These enhancements recognize advance care planning as an ongoing process of discussion and documentation that ensures a patient’s care and interventions reflect their values, beliefs, and preferences when unable to make decisions for themselves. It also emphasizes that goals of care are dynamic, and as they evolve, beneficiaries should feel supported and empowered to update their AHCDs affordably and with guidance from educational tools and trained professionals when needed.
Recommendation 3. Ensure electronic accessibility for healthcare providers.
CMS should also integrate the Medicare.gov AHCD storage system or a CMS-approved alternative with existing electronic health records (EHRs).
EHR systems in the United States currently lack full interoperability, meaning that when patients move through the continuum of care—from preventive services to medical treatment, rehabilitation, ongoing care maintenance—and between healthcare systems, their medical records, including AHCDs, may not transfer with them. This makes it challenging for healthcare providers to efficiently access these directives and deliver care that aligns with a patient’s wishes when the patient is incapacitated. To address this, CMS could encourage the integration of the Medicare.gov AHCD storage system or an alternative CMS-approved secure ACP digital platform to interface with all EHRs.
This storage platform could operate as an external add-on feature, allowing AHCDs to be accessible through any EHR, regardless of the healthcare system. Such external add-ons are typically third-party tools or modules that integrate with existing EHR systems to extend functionality, often addressing needs not covered by the core system. These add-ons are commonly used to connect EHRs with tools like clinical decision support systems, telehealth platforms, health information exchanges, and patient communication tools.
Such a universal, electronic system would prevent AHCDs from being misplaced, make them easily accessible across different states and health systems, and allow for easy updates. This would ensure that Medicare beneficiaries’ end-of-life care preferences are consistently honored, regardless of where they receive care.
Recommendation 4. Provide financial incentives for AHCD completion.
CMS should offer financial incentives for completing an AHCD, including options like tax credits, reduced or waived copayments and deductibles, prescription rebates, or other health-related subsidies.
Medicare’s increasing monthly premiums and cost-sharing requirements are often a substantial burden, especially for beneficiaries on fixed incomes. Nearly one in four Medicare enrollees age 65 and older report difficulty affording premiums, and almost 40% of those with incomes below twice the federal poverty level struggle to cover these costs. Additional financial burdens arise from extended care beyond standard coverage limits.
For example, in 2024, Medicare requires beneficiaries to pay $408 per day for inpatient, rehabilitation, inpatient psychiatric, and long-term acute care between days 61–90 of a hospital stay, totaling $11,832. Beyond 90 days, beneficiaries incur $816 per day for up to 60 lifetime reserve days, amounting to $48,720. Once these lifetime reserve days are exhausted, patients bear all inpatient costs, and these reserve days are never replenished again once they are used. Although the average hospital length of stay is typically shorter, inpatient days under Medicare do not need to be consecutive. This means if a patient is discharged and readmitted within a 60-day period, these patient payment responsibilities still apply and will not reset until there has been at least a 60-day break in care.
Medicare coverage for skilled nursing facilities is similarly limited: While Medicare fully covers the first 20 days when transferred from a qualified inpatient stay (at least three consecutive inpatient days, excluding the day of discharge), days 21–100 require a copayment of $204 per day, totaling $16,116. After 100 days, all SNF costs fall to the beneficiary. These costs are significant, and without out-of-pocket maximums, they can create financial hardship.
Some of these costs can be subsidized with Medicare Supplemental Insurance, or Medigap plans, but they come with additional premiums. By regularly educating patients and families of these costs—and offering tax credits, waived or reduced copayments and deductibles, prescription rebates, or account credits—CMS could provide substantial financial relief while encouraging the completion of AHCDs.
Encourage Expansion of the NCQA’s ACP HEDIS Measure
Finally, the MAHDE Initiative can be coupled with the expansion of the HEDIS measure to establish a comprehensive strategy for advancing proactive healthcare planning among Medicare beneficiaries. By encouraging both the accessibility and completion of AHCDs, while also integrating ACP as a quality measure for all Medicare Advantage enrollees aged 65 and older, CMS would embed ACP into standard patient care. This approach would incentivize health plans to prioritize ACP and help align patients’ care goals with the services they receive, fostering a more patient-centered, value-driven model of care within Medicare.
Figure 2. Four key features of the Medicare Advance Healthcare Directive Enrollment (MAHDE) Initiative. This initiative should also work in tandem with efforts to encourage the NCQA to expand ACP HEDIS measures to include all Medicare Advantage beneficiaries aged 65 and older. (Source: Dr. Tiffany Chioma Anaebere)
Conclusion
When patients and their families are clear on their goals of care, it is much less challenging for medical staff to navigate crises and stressful clinical situations. In unfortunate cases when these decisions have not been discussed and documented before a patient becomes incapacitated, doctors witness families struggle deeply with these choices, often leading to intense disagreements, conflict, and guilt. This uncertainty can also result in care that may not align with the patient’s goals.
Physicians and other qualified health professionals should continue to be trained on best practices to facilitate ACP with patients, and more importantly, the system should be redesigned to support these conversations early and often for all older Americans. The MAHDE Initiative is feasible, empowers patients to engage in ACP, and will reduce medical costs nationwide by allowing patients to be educated about their options and choose the care they want in future emergencies and at the end of life.
Starting an AHCD enrollment initiative with the Medicare population older than age 65 and achieving success in this group can pave the way for expanding ACP efforts to other high-need groups and, eventually, the general population. This approach fosters a healthcare environment where, as a nation, we become more comfortable discussing and managing healthcare decisions during emergencies and at the end of life.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
No. While the MAHDE Initiative will encourage all adults over age 65 who are enrolled in Medicare or Medicare Advantage plans to complete or renew an electronic AHCD annually through Medicare.gov or an alternative CMS-approved secure ACP digital platform, it will not be a requirement for receiving Medicare benefits or care.
Working alongside state-specific submission guidelines, Medicare beneficiaries can securely complete their AHCD on their own or during a visit with a qualified medical provider or health professional, either in person or through telehealth.
- Online submission: An accessible electronic version will be available on Medicare.gov or an alternative CMS-approved secure ACP digital platform, allowing individuals to complete and submit their AHCD online, with guidance from their care provider as needed.
- Paper version: Alternatively, individuals can also choose to complete a paper version of the AHCD, which can then be submitted to Medicare or a CMS-approved alternative for well-digitized electronic upload, storage, and access by healthcare professionals on Medicare.gov.
Review, updates, or to or confirm “no change” to these directives can be made annually online or by resubmitting updated paper forms during open enrollment or anytime as desired. Flexible options aim to make the process of AHCD completion accessible and convenient for all Medicare beneficiaries.
The author does not endorse specific products, individuals, or organizations. Any references are intended as examples or options for further exploration, not as endorsements or formal recommendations.
Driving Equitable Healthcare Innovations through an AI for Medicaid (AIM) Initiative
Artificial intelligence (AI) has transformative potential in the public health space – in an era when millions of Americans have limited access to high-quality healthcare services, AI-based tools and applications can enable remote diagnostics, drive efficiencies in implementation of public health interventions, and support clinical decision-making in low-resource settings. However, innovation driven primarily by the private sector today may be exacerbating existing disparities by training models on homogenous datasets and building tools that primarily benefit high socioeconomic status (SES) populations.
To address this gap, the Center for Medicare and Medicaid Innovation (CMMI) should create an AI for Medicaid (AIM) Initiative to distribute competitive grants to state Medicaid programs (in partnership with the private sector) for pilot AI solutions that lower costs and improve care delivery for rural and low-income populations covered by Medicaid.
Challenge & Opportunity
In 2022, the United States spent $4.5 trillion on healthcare, accounting for 17.3% of total GDP. Despite spending far more on healthcare per capita compared to other high-income countries, the United States has significantly worse outcomes, including lower life expectancy, higher death rates due to avoidable causes, and lesser access to healthcare services. Further, the 80 million low-income Americans reliant on state-administered Medicaid programs often have below-average health outcomes and the least access to healthcare services.
AI has the potential to transform the healthcare system – but innovation solely driven by the private sector results in the exacerbation of the previously described inequities. Algorithms in general are often trained on datasets that do not represent the underlying population – in many cases, these training biases result in tools and models that perform poorly for racial minorities, people living with comorbidities, and people of low SES. For example, until January 2023, the model used to prioritize patients for kidney transplants systematically ranked Black patients lower than White patients – the race component was identified and removed due to advocacy efforts within the medical community. AI models, while significantly more powerful than traditional predictive algorithms, are also more difficult to understand and engineer, resulting in the likelihood of further perpetuating such biases.
Additionally, startups innovating the digital health space today are not incentivized to develop solutions for marginalized populations. For example, in FY 2022, the top 10 startups focused on Medicaid received only $1.5B in private funding, while their Medicare Advantage (MA)-focused counterparts received over $20B. Medicaid’s lower margins are not attractive to investors, so digital health development targets populations that are already well-insured and have higher degrees of access to care.
The Federal Government is uniquely positioned to bridge the incentive gap between developers of AI-based tools in the private sector and American communities who would benefit most from said tools. Accordingly, the Center for Medicare and Medicaid Innovation (CMMI) should launch the AI for Medicaid (AIM) Initiative to incentivize and pilot novel AI healthcare tools and solutions targeting Medicaid recipients. Precedents in other countries demonstrate early success in state incentives unlocking health AI innovations – in 2023, the United Kingdom’s National Health Service (NHS) partnered with Deep Medical to pilot AI software that streamlines services by predicting and mitigating missed appointment risk. The successful pilot is now being adopted more broadly and is projected to save the NHS over $30M annually in the coming years.
The AIM Initiative, guided by the structure of the former Medicaid Innovation Accelerator Program (IAP), President Biden’s executive order on integrating equity into AI development, and HHS’ Equity Plan (2022), will encourage the private sector to partner with State Medicaid programs on solutions that benefit rural and low-income Americans covered by Medicaid and drive efficiencies in the overall healthcare system.
Plan of Action
CMMI will launch and operate the AIM Initiative within the Department of Health and Human Services (HHS). $20M of HHS’ annual budget request will be allocated towards the program. State Medicaid programs, in partnership with the private sector, will be invited to submit proposals for competitive grants. In addition to funding, CMMI will leverage the former structure of the Medicaid IAP program to provide state Medicaid agencies with technical assistance throughout their participation in the AIM Initiative. The programs ultimately selected for pilot funding will be monitored and evaluated for broader implementation in the future.
Sample Detailed Timeline
- 0-6 months:
- HHS Secretary to announce and launch the AI for Medicaid (AIM) Initiative within CMMI (e.g., delineating personnel responsibilities and engaging with stakeholders to shape the program)
- HHS to include AIM funding in annual budget request to Congress ($20M allocation)
- 6-12 months:
- CMMI to engage directly with state Medicaid agencies to support proposal development and facilitate connections with private sector partners
- CMMI to complete solicitation period and select ~7-10 proposals for pilot funding of ~$2-5M each by end of Year 1
- Year 2-7: Launch and roll out selected AI projects, led by state Medicaid agencies with continued technical assistance from CMMI
- Year 8: CMMI to produce an evaluative report and provide recommendations for broader adoption of AI tools and solutions within Medicaid-covered and other populations
Risks and Limitations
- Participation: Success of the initiative relies on state Medicaid programs and private sector partners’ participation. To mitigate this risk, CMMI will engage early with the National Association of Medicaid Directors (NAMD) to generate interest and provide technical assistance in proposal development. These conversations will also include input and support from the HHS Office of the Chief AI Officer (OCAIO) and its AI Council/Community of Practice. Further, startups in the healthcare AI space will be invited to engage with CMMI on identifying potential partnerships with state Medicaid agencies. A secondary goal of the initiative will be to ensure a number of private sector partners are involved in AIM.
- Oversight: AI is at the frontier of technological development today, and it is critical to ensure guardrails are in place to protect patients using AI technologies from potential adverse outcomes. To mitigate this risk, state Medicaid agencies will be required to submit detailed evaluation plans with their proposals. Additionally, informed consent and the ability to opt-out of data sharing when engaging with personally identifiable information (PII) and diagnostic or therapeutic technologies will be required. Technology partners (whether private, academic, or public sector) will further be required to demonstrate (1) adequate testing to identify and reduce bias in their AI tools to reasonable standards, (2) engagement with beneficiaries in the development process, and (3) leveraging testing environments that reflect the particular context of the Medicaid population. Finally, all proposals must adhere to guidelines published by AI guidelines adopted by HHS and the federal government more broadly, such as the CMS AI Playbook, the HHS Trustworthy AI Playbook, and any imminent regulations.
- Longevity: As a pilot grant program, the initiative does not promise long-term results for the broader population and will only facilitate short-term projects at the state level. Consequently, HHS leadership must remain committed to program evaluation and a long-term outlook on how AI can be integrated to support Americans more broadly. AI technologies or tools considered for acquisition by state Medicaid agencies or federal agencies after pilot implementation should ensure compliance with OMB guidelines.
Conclusion
The AI for Medicaid Initiative is an important step in ensuring the promise of artificial intelligence in healthcare extends to all Americans. The initiative will enable the piloting of a range of solutions at a relatively low cost, engage with stakeholders across the public and private sectors, and position the United States as a leader in healthcare AI technologies. Leveraging state incentives to address a critical market failure in the digital health space can additionally unlock significant efficiencies within the Medicaid program and the broader healthcare system. The rural and low-income Americans reliant on Medicaid have too often been an afterthought in access to healthcare services and technologies – the AIM Initiative provides an opportunity to address this health equity gap.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
Promoting Fairness in Medical Innovation
There is a crisis within healthcare technology research and development, wherein certain groups due to their age, gender, or race and ethnicity are under-researched in preclinical studies, under-represented in clinical trials, misunderstood by clinical practitioners, and harmed by biased medical technology. These issues in turn contribute to costly disparities in healthcare outcomes, leading to losses of $93 billion a year in excess medical-care costs, $42 billion a year in lost productivity, and $175 billion a year due to premature deaths. With the rise of artificial intelligence (AI) in healthcare, there’s a risk of encoding and recreating existing biases at scale.
The next Administration and Congress must act to address bias in medical technology at the development, testing and regulation, and market-deployment and evaluation phases. This will require coordinated effort across multiple agencies. In the development phase, science funding agencies should enforce mandatory subgroup analysis for diverse populations, expand funding for under-resourced research areas, and deploy targeted market-shaping mechanisms to incentivize fair technology. In the testing and regulation phase, the FDA should raise the threshold for evaluation of medical technologies and algorithms and expand data-auditing processes. In the market-deployment and evaluation phases, infrastructure should be developed to perform impact assessments of deployed technologies and government procurement should incentivize technologies that improve health outcomes.
Challenge and Opportunity
Bias is regrettably endemic in medical innovation. Drugs are incorrectly dosed to people assigned female at birth due to historical exclusion of women from clinical trials. Medical algorithms make healthcare decisions based on biased health data, clinically disputed race-based corrections, and/or model choices that exacerbate healthcare disparities. Much medical equipment is not accessible, thus violating the Americans with Disabilities Act. And drugs, devices, and algorithms are not designed with the lifespan in mind, impacting both children and the elderly. Biased studies, technology, and equipment inevitably produce disparate outcomes in U.S. healthcare.
The problem of bias in medical innovation manifests in multiple ways: cutting across technological sectors in clinical trials, pervading the commercialization pipeline, and impeding equitable access to critical healthcare advances.
Bias in medical innovation starts with clinical research and trials
The 1993 National Institutes of Health (NIH) Revitalization Act required federally funded clinical studies to (i) include women and racial minorities as participants, and (ii) break down results by sex and race or ethnicity. As of 2019, the NIH also requires inclusion of participants across the lifespan, including children and older adults. Yet a 2019 study found that only 13.4% of NIH-funded trials performed the mandatory subgroup analysis, and challenges in meeting diversity targets continue into 2024 . Moreover, the increasing share of industry-funded studies are not subject to Revitalization Act mandates for subgroup analysis. These studies frequently fail to report differences in outcomes by patient population as a result. New requirements for Diversity Action Plans (DAPs), mandated under the 2023 Food and Drug Omnibus Reform Act, will ensure drug and device sponsors think about enrollment of diverse populations in clinical trials. Yet, the FDA can still approve drugs and devices that are not in compliance with their proposed DAPs, raising questions around weak enforcement.
The resulting disparities in clinical-trial representation are stark: African Americans represent 12% of the U.S. population but only 5% of clinical-trial participants, Hispanics make up 16% of the population but only 1% of clinical trial participants, and sex distribution in some trials is 67% male. Finally, many medical technologies approved prior to 1993 have not been reassessed for potential bias. One outcome of such inequitable representation is evident in drug dosing protocols: sex-aware prescribing guidelines exist for only a third of all drugs.
Bias in medical innovation is further perpetuated by weak regulation
Algorithms
Regulation of medical algorithms varies based on end application, as defined in the 21st Century Cures Act. Only algorithms that (i) acquire and analyze medical data and (ii) could have adverse outcomes are subject to FDA regulation. Thus, clinical decision-support software (CDS) is not regulated even though these technologies make important clinical decisions in 90% of U.S. hospitals. The FDA has taken steps to try and clarify what CDS must be considered a medical device, although these actions have been heavily criticized by industry. Finally, the lack of regulatory frameworks for generative AI tools is leading to proliferation without oversight.
Even when a medical algorithm is regulated, regulation may occur through relatively permissive de novo pathways and 510(k) pathways. A de novo pathway is used for novel devices determined to be low to moderate risk, and thus subject to a lower burden of proof with respect to safety and equity. A 510(k) pathway can be used to approve a medical device exhibiting “substantial equivalence” to a previously approved device, i.e., it has the same intended use and/or same technological features. Different technical features can be approved so long as there are no questions raised around safety and effectiveness.
Medical algorithms approved through de novo pathways can be used as predicates for approval of devices through 510(k) pathways. Moreover, a device approved through a 510(k) pathway can remain on the market even if its predicate device was recalled. Widespread use of 510(k) approval pathways has generated a “collapsing building” phenomenon, wherein many technologies currently in use are based on failed predecessors. Indeed, 97% of devices recalled between 2008 to 2017 were approved via 510(k) clearance.
While DAP implementation will likely improve these numbers, for the 692 AI-ML enabled medical devices, only 3.6% reported race or ethnicity, 18.4% reported age, and only .9% include any socioeconomic information. Further, less than half did detailed analysis of algorithmic performance and only 9% included information on post-market studies, raising the risk of algorithmic bias following approvals and broad commercialization.
Even more alarming is evidence showing that machine learning can further entrench medical inequities. Because machine learning medical algorithms are powered by data from past medical decision-making, which is rife with human error, these algorithms can perpetuate racial, gender, and economic bias. Even algorithms demonstrated to be ‘unbiased’ at the time of approval can evolve in biased ways over time, with little to no oversight from the FDA. As technological innovation progresses, especially generative AI tools, an intentional focus on this problem will be required.
Medical devices
Currently, the Medical Device User Fee Act requires the FDA to consider the least burdensome appropriate means for manufacturers to demonstrate the effectiveness of a medical device or to demonstrate a device’s substantial equivalence. This requirement was reinforced by the 21st Century Cures Act, which also designated a category for “breakthrough devices” subject to far less-stringent data requirements. Such legislation shifts the burden of clinical data collection to physicians and researchers, who might discover bias years after FDA approval. This legislation also makes it difficult to require assessments on the differential impacts of technology.
Like medical algorithms, many medical devices are approved through 510(k) exemptions or de novo pathways. The FDA has taken steps since 2018 to increase requirements for 510(k) approval and ensure that Class III (high-risk) medical devices are subject to rigorous pre-market approval, but problems posed by equivalence and limited diversity requirements remain.
Finally, while DAPs will be required for many devices seeking FDA approval, the recommended number of patients in device testing is shockingly low. For example, currently, only 10 people are required in a study of any new pulse oximeter’s efficacy and only 2 of those people need to be “darkly pigmented”. This requirement (i) does not have the statistical power necessary to detect differences between demographic groups, and (i) does not represent the composition of the U.S. population. The standard is currently under revision after immense external pressure. FDA-wide, there are no recommended guidelines for addressing human differences in device design, such as pigmentation, body size, age, and pre-existing conditions.
Pharmaceuticals
The 1993 Revitalization Act strictly governs clinical trials for pharmaceuticals and does not make recommendations for adequate sex or genetic diversity in preclinical research. The results are that a disproportionately high number of male animals are used in research and that only 5% of cell lines used for pharmaceutical research are of African descent. Programs like All of Us, an effort to build diverse health databases through data collection, are promising steps towards improving equity and representation in pharmaceutical research and development (R&D). But stronger enforcement is needed to ensure that preclinical data (which informs function in clinical trials) reflects the diversity of our nation.
Bias in medical innovation are not tracked post-regulatory approval
FDA-regulated medical technologies appear trustworthy to clinicians, where the approval signals safety and effectiveness. So, when errors or biases occur (if they are even noticed), the practitioner may blame the patient for their lifestyle rather than the technology used for assessment. This in turn leads to worse clinical outcomes as a result of the care received.
Bias in pulse oximetry is the perfect case study of a well-trusted technology leading to significant patient harm. During the COVID-19 pandemic, many clinicians and patients were using oximeter technology for the first time and were not trained to spot factors, like melanin in the skin, that cause inaccurate measurements and impact patient care. Issues were largely not attributed to the device. This then leads to underreporting of adverse events to the FDA — which is already a problem due to the voluntary nature of adverse-event reporting.
Even when problems are ultimately identified, the federal government is slow to respond. The pulse oximeter’s limitations in monitoring oxygenation levels across diverse skin tones was identified as early as the 1990s. 34 years later, despite repeated follow-up studies indicating biases, no manufacturer has incorporated skin-tone-adjusted calibration algorithms into pulse oximeters. It required the large Sjoding study, and the media coverage it garnered around delayed care and unnecessary deaths, for the FDA to issue a safety communication and begin reviewing the regulation.
Other areas of HHS are stepping up to address issues of bias in deployed technologies. A new ruling by the HHS Office of Civil Rights (OCR) on Section 1557 of the Affordable Care Act requires covered providers and institutions (i.e. any receiving federal funding) to identify their use of patient care decision support tools that directly measure race, color, national origin, sex, age, or disability, and to make reasonable efforts to mitigate the risk of discrimination from their use of these tools. Implementation of this rule will depend on OCR’s enforcement, and yet it provides another route to address bias in algorithmic tools.
Differential access to medical innovation is a form of bias
Americans face wildly different levels of access to new medical innovations. As many new innovations have high cost points, these drugs, devices, and algorithms exist outside the price range of many patients, smaller healthcare institutions and federally funded healthcare service providers, including the Veterans Health Administration, federally qualified health centers and the Indian Health Service. Emerging care-delivery strategies might not be covered by Medicare and Medicaid, meaning that patients insured by CMS cannot access the most cutting-edge treatments. Finally, the shift to digital health, spurred by COVID-19, has compromised access to healthcare in rural communities without reliable broadband access.
Finally, the Advanced Research Projects Agency for Health (ARPA-H) has a commitment to have all programs and projects consider equity in their design. To fulfill ARPA-H’s commitment, there is a need for action to ensure that medical technologies are developed fairly, tested with rigor, deployed safely, and made affordable and accessible to everyone.
Plan of Action
The next Administration should launch “Healthcare Innovation for All Americans” (HIAA), a whole of government initiative to improve health outcomes by ensuring Americans have access to bias-free medical technologies. Through a comprehensive approach that addresses bias in all medical technology sectors, at all stages of the commercialization pipeline, and in all geographies, the initiative will strive to ensure the medical-innovation ecosystem works for all. HIAA should be a joint mandate of Health and Human Services (HHS) and the Office of Science Technology and Policy (OSTP) to work with federal agencies on priorities of equity, non-discrimination per Section 1557 of the Affordable Care Act and increasing access to medical innovation, and initiative leadership should sit at both HHS and OSTP.
This initiative will require involvement of multiple federal agencies, as summarized in the table below. Additional detail is provided in the subsequent sections describing how the federal government can mitigate bias in the development phase; testing, regulation, and approval phases; and market deployment and evaluation phases.
Three guiding principles should underlie the initiative:
- Equity and non-discrimination should drive action. Actions should seek to improve the health of those who have been historically excluded from medical research and development. We should design standards that repair past exclusion and prevent future exclusion.
- Coordination and cooperation are necessary. The executive and legislative branches must collaborate to address the full scope of the problem of bias in medical technology, from federal processes to new regulations. Legislative leadership should task the Government Accountability Office (GAO) to engage in ongoing assessment of progress towards the goal of achieving bias-free and fair medical innovation.
- Transparent, evidence-based decision making is paramount. There is abundant peer-reviewed literature that examines bias in drugs, devices, and algorithms used in healthcare settings — this literature should form the basis of a non-discrimination approach to medical innovation. Gaps in evidence should be focused on through deployed research funding. Moreover, as algorithms become ubiquitous in medicine, every effort should be made to ensure that these algorithms are trained on representative data of those experiencing a given healthcare condition.
Addressing bias at the development phase
The following actions should be taken to address bias in medical technology at the innovation phase:
- Enforce parity in government-funded research. For clinical research, NIH should examine the widespread lack of adherence to regulations requiring that government funded clinical trials report sex, racial or ethnicity, and age breakdown of trial participants. Funding should be reevaluated for non-compliant trials. For preclinical research, NIH should require gender parity in animal models and representation of diverse cell lines used in federally funded studies.
- Deploy funding to address research gaps. Where data sources for historically marginalized people are lacking, such as for women’s cardiovascular health, NIH should deploy strategic, targeted funding programs to fill these knowledge gaps. This could build on efforts like the Initiative on Women’s Health Research. Increased funding should include resources for underrepresented groups to participate in research and clinical trials through building capacity in community organizations. Results should be added to a publicly available database so they can be accessed by designers of new technologies. Funding programs should also be created to fill gaps in technology, such as in diagnostics and treatments for high-prevalence and high-burden uterine diseases like endometriosis (found in 10% of reproductive-aged people with uteruses).
- Invest in research into healthcare algorithms and databases. Given the explosion of algorithms in healthcare decision-making, NIH and NSF should launch a new research program focused on the study, evaluation, and application of algorithms in healthcare delivery, and on how artificial intelligence and machine learning (AI/ML) can exacerbate healthcare inequities. The initial request for proposals should focus on design strategies for medical algorithms that mitigate bias from data or model choices.
- Task ARPA-H with developing metrics for equitable medical technology development. ARPA-H should prioritize developing a set of procedures and metrics for equitable development of medical technology. Once developed, these processes should be rapidly deployed across ARPA-H, as well as published for potential adoption by additional federal agencies, industry, and other stakeholders. ARPA-H could also collaborate with NIST on standards setting with NIST and ASTP on relevant standards setting. For instance, NIST has developed an AI Risk Management Framework and the ONC engages in setting standards that achieve equity by design. CMS could use resultant standards for Medicare and Medicaid reimbursements.
- Leverage procurement as a demand-signal for medical technologies that work for diverse populations. As the nation’s largest healthcare system, the Veterans Health Administration (VHA) can generate demand-signals for bias-free medical technologies through its procurement processes and market-shaping mechanisms. For example, the VA could put out a call for a pulse oximeter that works equally well across the entire range of human skin pigmentation and offer contracts for the winning technology.
Addressing bias at the testing, regulation, and approval phases
The following actions should be taken to address bias in medical innovation at the testing, regulation, and approval phases:
- Raise the threshold for FDA evaluation of devices and algorithms. Equivalency necessary to receive 510(k) clearance should be narrowed. For algorithms, this would involve consideration of whether the datasets for machine learning tactics used by the new device and its predicate are similar. For devices (including those that use algorithms), this would require tightening the definition of “same intended use” (currently defined as a technology having the same functionality as one previously approved by the FDA) as well as eliminating the approval of new devices with “different technological characteristics” (the application of one technology to a new area of treatment in which that technology is untested).
- Evaluate FDA’s guidance on specific technology groups for equity. Requirements for the safety of a given drug, medical device, or algorithm should have the statistical power necessary to detect differences between demographic groups and represent all end-users of the technology..
- Establish a data bank for auditing medical algorithms. The newly established Office of Digital Transformation within the FDA should create a “data bank” of healthcare images and datasets representative of the U.S. population, which could be done in partnership with the All of Us program. Medical technology developers could use the data bank to assess the performance of medical algorithms across patient populations. Regulators could use the data bank to ground claims made by those submitting a technology for FDA approval.
- Allow data submitted to the FDA to be examined by the broader scientific community. Currently, data submitted to the FDA as part of its regulatory-approval process is kept as a trade secret and not released pre-authorization to researchers. Releasing the data via an FDA-invited “peer review” step in the regulation of high-risk technologies, like automated decision-making algorithms, Class III medical devices, and drugs, will ensure that additional, external rigor is applied to the technologies that could cause the most harm due to potential biases.
- Establish an enforceable AI Bill of Rights. The federal government and Congress should create protections for necessary uses of artificial intelligence (AI) identified by OSTP. Federally funded healthcare centers, like facilities part of the Veterans Health Administration, could refuse to buy software or technology products that violate this “AI Bill of Rights” through changes to federal acquisition regulation (FAR).
Addressing bias at the market deployment and evaluation phases
- Strengthen reporting mechanisms at the FDA. Healthcare providers, who are often closest to the deployment of medical technologies, should be made mandatory reporters to the FDA of all witnessed adverse events related to bias in medical technology. In addition, the FDA should require the inclusion of unique device identifiers (UDIs) in adverse-response reporting. Using this data, Congress should create a national and publicly accessible registry that uses UDIs to track post-market medical outcomes and safety.
- Require impact assessments of deployed technologies. Congress must establish systems of accountability for medical technologies, like algorithms, that can evolve over time. Such work could be done by passing the Algorithmic Accountability Act which would require companies that create “high-risk automated decision systems” to conduct impact assessments reviewed by the FTC as frequently as necessary.
- Assess disparities in patient outcomes to direct technical auditing. AHRQ should be given the funding needed to fully investigate patient-outcome disparities that could be caused by biases in medical technology, such as its investigation into the impacts of healthcare algorithms on racial and ethnic disparities. The results of this research should be used to identify technologies that the FDA should audit post-market for efficacy or the FTC should investigate. CMS and its accrediting agencies can monitor these technologies and assess whether they should receive Medicare and Medicaid funding.
- Review reimbursement guidelines that are dependent on medical technologies with known bias. CMS should review its national coverage determinations for technologies, like pulse oximetry, that are known to perform differently across populations. For example, pulse oximeters can be used to determine home oxygen therapy provision, thus potentially excluding darkly-pigmented populations from receiving this benefit.
- Train physicians to identify bias in medical technologies and identify new areas of specialization. ED should work with medical schools to develop curricula training physicians to identify potential sources of bias in medical technologies and ensuring that physicians understand how to report adverse events to the FDA. In addition, ED should consider working with the American Medical Association to create new medical specialties that work at the intersection of technology and care delivery.
- Ensure that technologies developed by ARPA-H have an enforceable access plan. ARPA-H will produce cutting-edge technologies that must be made accessible to all Americans. ARPA-H should collaborate with the Center for Medicare and Medicaid Innovation to develop strategies for equitable delivery of these new technologies. A cost-effective deployment strategy must be identified to service federally-funded healthcare institutions like Veterans Health Administration hospitals and clinical, federally qualified health centers, and Indian Health Service.
- Create a fund to support digital health technology infrastructure in rural hospitals. To capitalize on the $65 billion expansion of broadband access allocated in the Bipartisan Infrastructure Bill, HRSA should deploy strategic funding to federally qualified health centers and rural health clinics to support digital health strategies — such as telehealth and mobile health monitoring — and patient education for technology adoption.
A comprehensive road map is needed
The GAO should conduct a comprehensive investigation of “black box” medical technologies utilizing algorithms that are not transparent to end users, medical providers, and patients. The investigation should inform a national strategic plan for equity and non-discrimination in medical innovation that relies heavily on algorithmic decision-making. The plan should include identification of noteworthy medical technologies leading to differential healthcare outcomes, creation of enforceable regulatory standards, development of new sources of research funding to address knowledge gaps, development of enforcement mechanisms for bias reporting, and ongoing assessment of equity goals.
Timeline for action
Realizing HIAA will require mobilization of federal funding, introduction of regulation and legislation, and coordination of stakeholders from federal agencies, industry, healthcare providers, and researchers around a common goal of mitigating bias in medical technology. Such an initiative will be a multi-year undertaking and require funding to enact R&D expenditures, expand data capacity, assess enforcement impacts, create educational materials, and deploy personnel to staff all the above.
Near-term steps that can be taken to launch HIAA include issuing a public request for information, gathering stakeholders, engaging the public and relevant communities in conversation, and preparing a report outlining the roadmap to accomplishing the policies outlined in this memo.
Conclusion
Medical innovation is central to the delivery of high-quality healthcare in the United States. Ensuring equitable healthcare for all Americans requires ensuring that medical innovation is equitable across all sectors, phases, and geographies. Through a bold and comprehensive initiative, the next Administration can ensure that our nation continues leading the world in medical innovation while crafting a future where healthcare delivery works for all.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
HIAA will be successful when medical policies, projects, and technologies yield equitable health care access, treatment, and outcomes. For instance, success would yield the following outcomes:
- Representation in preclinical and clinical research equivalent to the incidence of a studied condition in the general population.
- Research on a disease condition funded equally per affected patient.
- Existence of data for all populations facing a given disease condition.
- Medical algorithms that have equal efficacy across subgroup populations.
- Technologies that work equally well in testing as they do when deployed to the market.
- Healthcare technologies made available and affordable to all care facilities.
Regulation alone cannot close the disparity gap. There are notable gaps in preclinical and clinical research data for women, people of color, and other historically underrepresented groups that need to be filled. There are also historical biases encoded in AI/ML decision making algorithms that need to be studied and rectified. In addition, the FDA’s role is to serve as a safety check on new technologies — the agency has limited oversight over technologies once they are out on the market due to the voluntary nature of adverse reporting mechanisms. This means that agencies like the FTC and CMS need to be mobilized to audit high-risk technologies once they reach the market. Eliminating bias in medical technology is only possible through coordination and cooperation of federal agencies with each other as well as with partners in the medical device industry, the pharmaceutical industry, academic research, and medical care delivery.
A significant focus of the medical device and pharmaceutical industries is reducing the time to market for new medical devices and drugs. Imposing additional requirements for subgroup analysis and equitable use as part of the approval process could work against this objective. On the other hand, ensuring equitable use during the development and approval stages of commercialization will ultimately be less costly than dealing with a future recall or a loss of Medicare or Medicaid eligibility if discriminatory outcomes are discovered.
Healthcare disparities exist in every state in America and are costing billions a year in economic growth. Some of the most vulnerable people live in rural areas, where they are less likely to receive high-quality care because costs of new medical technologies are too high for the federally qualified health centers that serve one in five rural residents as well as rural hospitals. Furthermore, during continued use, a biased device creates adverse healthcare outcomes that cost taxpayers money. A technology functioning poorly due to bias can be expensive to replace. It is economically imperative to ensure technology works as expected, as it leads to more effective healthcare and thus healthier people.
Establish Data Standards To Protect Newborn DNA Privacy by Developing Data Storage Standards for Newborn Screening Samples
Newborn screening is performed on millions of babies in the U.S. every year to test for rare genetic diseases and, when necessary, allow for early treatment. While newborn screening is mandated by the federal government, each state runs its own screening program. Importantly, individual states manage how newborn screening data is stored and, potentially, accessed and used in the future. While such data is often used for quality assurance testing and clinical research, there have been instances of law enforcement subpoenaing newborn screening data for use in criminal investigations. For example, New Jersey used newborn screening data to investigate a decades-old sexual assault This raises major concerns about overall transparency of data use and privacy in the newborn screening process.
The incoming administration should encourage states to develop data handling standards for newborn screening data. Specifically these standards should include how long data is stored and who can access it. This can be accomplished by directing the Health and Human Services’ (HHS) Federal Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC) to provide recommendations that clearly communicate data use and privacy measures to state health departments. In addition, the incoming administration should also encourage development of increased educational materials for parents to explain these privacy concerns, and create funding opportunities to incentivize both of these measures.
Challenge and Opportunity
Newborn screening is a universal practice across the United States. Blood samples are taken from infants only a few days old to test for a variety of genetic diseases such as phenylketonuria, which can cause intellectual disability that can be prevented through changes in diet—if it is caught early enough. These blood samples can be used for both metabolic and genetic tests, depending on which disease is being tested for and how it is detected. Phenylketonuria, for example, is detected by high levels of a molecule called phenylalanine in the blood, while spinal muscular atrophy (SMA) is detected by changes in the genetic sequence of the gene associated with SMA. Newborn screening is an essential practice that identifies a wide range of severe diseases before symptoms occur, and three babies out of every 1,000 are identified with a genetic condition.
While newborn screening is required by the federal government, each state can determine which panel of diseases are tested. The Department of Health and Human Services established an Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC), which regularly updates a Recommended Uniform Screening Panel (RUSP) with conditions. For example, SMA was approved to the RUSP in 2018, and all 50 states have now added SMA to their screening panels. Much of the effort to both nominate conditions to the federal RUSP and to encourage individual states to adapt spinal muscular atrophy testing was led by patient advocacy groups such as CureSMA, and these sorts of groups play a significant role in the addition of future conditions. Similar efforts are underway for Krabbe disease, which was added to the RUSP in 2024 and is currently screened for in only twelve states, a number that may increase in the coming years as more states consider adding it to their panels. State advisory boards will review new disease nominations and, along with their status on the RUSP, will often consider how prevalent a disease is, if there are treatments available for the disease, and cost-effectiveness of screening for this condition. Regardless of which tests are performed, every state participates in newborn screening. Importantly, newborn screening does not require affirmative consent from parents—some states offer opt-out options, generally for religious reasons, but 98% of infants are screened.
Mandatory newborn screening programs have led health departments across the country to obtain genetic data from nearly every child in the country for decades. With recent developments in genetic sequencing technologies, this means that, theoretically, this newborn screening data could be repurposed for other functions. In 2022, the New Jersey Office of the Public Defender filed a lawsuit against the New Jersey Department of Health for complying with a subpoena to provide newborn screening data to the police as part of a sexual assault investigation. Specifically, law enforcement subpoenaed the blood sample of a suspect’s child, which they used to perform new DNA analysis to match DNA crime scene evidence. The lawsuit reveals that the New Jersey Department of Health has retained newborn screening blood spots for over twenty years; that the data obtained from the subpoena was used to bring criminal charges for a crime committed in 1996; and that the Office of the Public Defender were not provided information about how many similar subpoenas have been complied with in the past.
This case highlights the bigger issue of newborn screening data as the United States’ “hidden national DNA database.” Law enforcement has potential access to decades of samples that can be used for genetic analysis that were not intended for law enforcement use. Police in other states like California have also sought access to newborn screening databases for investigational purposes. In California, the state health department keeps samples indefinitely, and not only is this information not disclosed to parents, it no longer provides parents with an opt-out option. As law enforcement agencies across states begin to understand the magnitude of data that can be found in these databases, it is becoming clear that health department policies for regulating access to these data are lacking.
Using genetic data in law enforcement has become increasingly common. The practice of “investigative genetic genealogy,” or IGG, has made national headlines in recent years, in which law enforcement can access genetic data from publicly available databases to use in criminal investigations. These databases are full of genetic data that consumers who participate in direct-to-consumer genetic testing, such as 23andMe, can use to voluntarily upload and share their data with more people. IGG presents its own privacy concerns, but it is important to recognize the voluntary nature of both (a) participating in direct-to-consumer testing and (b) uploading it to a third-party website. Newborn screening, on the other hand, is not an optional practice.
Proponents of IGG argue that using genetic data is very effective at not only catching killers—and doing so quicker than without DNA data—but also exonerating innocents. However, this fact does not outweigh the major issues of privacy, transparency, and the fact that this approach potentially violates the fourth amendment’s protections against unreasonable search and seizures—especially when it comes to incorporating newborn screening data into these approaches. A previous court case found a hospital in violation of the fourth amendment for providing law enforcement with warrantless drug screening results from pregnant women, even though the women were under the impression they were receiving diagnostic tests. The Supreme Court argued that the hospital’s actions break down public trust in the health system, as patients have a “reasonable expectation of privacy” regarding their test results. While cases of subpoenaing newborn screening data may not currently violate any legal procedure, allowing law enforcement access to these data for use in future investigations, particularly without informing the individuals or parents involved, may also erode trust in the health system. This may lead to parents—when given the option—to opting out of newborn screening programs more often, leading to an increase in genetic and metabolic disorders going undiagnosed in newborns and causing major health problems in the future. In addition, with many scientists advocating for adopting whole-genome sequencing of newborns—instead of simply sequencing a panel of genes that are commonly identified as disease-causing in newborns—the amount of potential available genetic data could be staggering. As a result, the incoming administration needs to take action to address the lack of transparent policies regarding newborn screening data in order to maintain its success as a public health measure.
Plan of Action
Current genetic privacy legislation
The landscape of genetic privacy legislation is, currently, somewhat patchwork. At the federal level, the most relevant legislation includes (1) the Genetic Information Nondiscrimination Act (GINA), (2) the Affordable Care Act (ACA), and (3) the Health Insurance Portability and Accountability Act (HIPAA). GINA specifically prohibits genetic discrimination in health insurance and in the workplace. This means that health insurers cannot deny coverage based on genetic data, and employers cannot make hiring, firing, or promotion decisions based on genetic data. The ACA strengthens GINA’s stipulation against genetic discrimination in health insurance by mandating that any health insurance issuer must provide coverage to whomever applies, as well as including genetic information on the list of factors that cannot be considered when determining overage or premium costs. HIPAA additionally regulates genetic data gathered in a healthcare setting, which includes newborn screening data, but HIPAA-protected information can be shared at the request of a court order or subpoena. The FBI developed an interim policy regarding all types of forensic genetic genealogy—often used with direct-to-consumer genetic tests but could also be applicable to newborn screening—which states the criteria required for investigators to use this approach. Criteria includes the requirement that a case must be an unsolved violent crime. In addition, the interim policy states that investigative agencies must identify themselves as law enforcement—a previous case was solved by accessing genetic databases without disclosing this information to the database—and that any collected data must be destroyed upon conclusion of the case.
Additionally, many states have additional laws that strengthen genetic privacy regulation on top of federal regulations. Maryland passed a bill that regulates the use of genetic data in criminal investigations—specifically, it requires that law enforcement obtains informed consent from non-suspects before using their DNA in investigations. Other recent state regulations that address law enforcement access to genetic data in one way or another include Montana, which requires government agencies to obtain a warrant to access genetic data, and Tennessee, which explicitly allows law enforcement to access genetic data as long as they obtain a warrant or subpoena. Importantly, many of these laws are geared more towards addressing genetic data from direct-to-consumer testing and do not directly apply to newborn screening. Like federal legislation, state genetic privacy legislation is largely lacking in policies to address the use of newborn screening by law enforcement.
On top of legislation regarding genetic privacy, states all have their own respective policies regarding newborn screening that vary dramatically. For example, a court in Minnesota found that nonconsensual storage of newborn screening data for use outside of genetic screening purposes violates the state genetic privacy law stating that genetic information can only be distributed with an individual’s written consent, leading to Minnesota destroying its newborn screening samples. Other states have no legislation at all. Additionally, states can have laws addressing other, non-law enforcement uses of newborn screening data; another major use of newborn screening data is research.
Policy Recommendations
The incoming administration should address the lack of transparency in newborn screening data management by implementing the following recommendations:
Direct the ACHDNC to develop national recommendations detailing standards for newborn genetic screening sample and data handling.
These standards should include:
Standards for what the data can be used for outside of newborn screening, and by whom. Newborn screening data is used in additional ways outside of law enforcement; it can also be used for quality assurance to help ensure tests are working properly, to help develop new tests, and in clinical trials. There are compelling arguments for these uses; for clinical research, for example, this data can contribute towards research studying the disease the child may have been diagnosed with. However, for the sake of transparency, policy should state specifically what newborn genetic data can and cannot be used for, and who is allowed access to the data under these circumstances. For instance, Michigan has a program called the Michigan BioTrust, which takes the leftover, de-identified newborn screening samples for use in research towards understanding disease. Parents can choose to opt in or out at the time of screening, and parents—as well as children, upon turning 18—can change their mind and have their data removed later if they so choose. Regardless of state decisions on whether law enforcement should be able to access their newborn screening data, clearly stating what the data can be used for overall is paramount for parents to understand what happens to their children’s samples.
The length of time that blood samples and genetic data can be stored in state databases, and when, if ever, the data will be destroyed. As detailed by the lawsuit, New Jersey had been storing samples for over twenty years, although parents were not actually aware of this fact until the lawsuit was filed; potentially in response to this lawsuit, starting in November 2024, New Jersey will be destroying blood spots older than 2 years. Similarly, Delaware stores blood spot samples for three years before destroying them. While there is no definitive answer to what the best timeline for saving samples is, establishing a transparent timeline for how long samples can be stored in each state will improve data handling transparency.
What say, if any, do parents have in what is done with their child’s samples and data. In Texas, after a lawsuit determining that storing newborn screening samples without consent was against the law, parents have the right to request their child’s samples be destroyed if they so choose. Developing policies that allow parents—or children themselves, once they become adults—to have a say in what happens to the samples after screening is completed would provide individuals control of their data without disincentivizing testing.
Partner with state advisory boards to develop educational materials for parents detailing ACHDNC recommendations and state-specific policy.
While newborn screening is mandated, there is variable information available to parents regarding what is done with the data. For example, Michigan has an extensive Q&A page on their Department of Health website addressing many major newborn screening-related questions, including a section addressing what is done with samples after screening is complete. In contrast, West Virginia’s Q&A page does not address what happens to the samples after testing. Not only would developing standard policies for data handling be beneficial, but improving the dissemination of such information to parents would increase overall transparency and improve trust in the system. The incoming administration should work closely with state advisory boards to improve the communication of newly-developed data handling standards to parents and other relevant parties.
Incentivize development of plans by providing grant opportunities to state health departments to support newborn screening programs.
Currently, newborn screening programs receive no direct federal funding; however, costs include operating costs, testing equipment, and personnel on top of the tests themselves. In general, newborn screening is paid for through a fee for the tests, which are often covered by the parents’ health insurance, or the State Children’s Health Insurance Program or Medicaid. However, grants such as the NBS Co-Propel have been awarded to states in the past for creating improvements in their newborn screening programs such as support for long-term follow up on patients that have positive test results returned to them. The Co-Propel grant was administered through the Maternal & Child Health Bureau (MCHB) of Health and Human Services; the incoming administration could recommend that MCHB initiates a new funding opportunity for states to either develop data storage standards and/or educational materials for families to encourage the adaptation of these standards.
Conclusion
Newborn genetic screening is an essential public health measure that saves thousands of lives each year by identifying diseases in newborns that can either be prevented early or treated immediately rather than waiting until severe symptom onset. However, with the advent of new genetic technologies and the burgeoning use of newborn genetic screening data in law enforcement investigations, major privacy and transparency issues are becoming known to parents, potentially putting trust in the newborn screening process at risk. This could reduce desire to participate in these programs, leading to an inability to quickly diagnose many preventable or treatable conditions. The incoming administration should work towards encouraging state health departments to develop clear and well-communicated data storage standards for newborn screening samples in order to combat these concerns moving forward.
This action-ready policy memo is part of Day One 2025 — our effort to bring forward bold policy ideas, grounded in science and evidence, that can tackle the country’s biggest challenges and bring us closer to the prosperous, equitable and safe future that we all hope for whoever takes office in 2025 and beyond.
PLEASE NOTE (February 2025): Since publication several government websites have been taken offline. We apologize for any broken links to once accessible public data.
Newborn screening is performed by pricking a newborn’s heel to obtain a blood sample, or “blood spot,” within two days of being born. These blood spot samples are used for both metabolic tests and genetic tests. Metabolic tests measure different molecules in the blood that might signal a disease, such as high levels of an amino acid called phenylalanine, which in healthy amounts is used by our bodies to make proteins and in high amounts can cause phenylketonuria. Genetic tests are performed by sequencing a panel, or selection, of genes that are often associated with newborn screening diagnoses; often, genetic testing is performed after a positive hit on a metabolic test to both confirm and further clarify the diagnosis.
The role of the Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC) is to communicate with the Secretary of the Department of Health and Human Services regarding newborn screening policies. This not only includes managing the Recommended Uniform Screening Panel, but also providing advice on grants and research projects related to newborn screening research, assistance with developing policies for state and local health departments for newborn screening implementation, and recommendations towards reducing child mortality from the diseases screened.
The Recommended Uniform Screening Panel (RUSP) is the list of disorders recommended for newborn testing. As of July 2024, the RUSP contains 38 “core conditions,” which are conditions that states specifically test for, and 26 “secondary conditions,” which are conditions that physicians may identify incidentally while screening for core conditions. New conditions can be added, and conditions can be moved between categories if the advisory board chooses to do so. These conditions include metabolic disorders such as phenylketonuria, endocrine disorders such as thyroid disorders, hemoglobin disorders such as sickle cell anemia, and others such as cystic fibrosis.
The National Human Genome Research Institute has a searchable database that details the different state genetic privacy laws, including their legislative status and a summary of their intended purpose. These laws have many goals, including expanding protections against genetic discrimination, research subject protections, artificial intelligence, and more.
Improving Public Awareness and Understanding of Advisory Committees
From January 2024 to July 2024, the Federation of American Scientists interviewed 30 current and former Advisory Committee (AdComm) members. Based on these discussions, we were able to source potential policy recommendations that may assist with enhancing the FDA’s ability to obtain valuable advice for evidence-based decision-making. The results of these discussions are presented in case study format detailing the recurring themes that emerged and policy recommendations for improvement.
The FDA holds one of the most important roles as a federal agency which is to ensure public safety when approving vaccines, medical devices, and medicines. The approval of these products usually require extensive trials with data that supports their safety and efficacy. Considering that most of these decisions are complex and multifaceted, the FDA enlists the support of Advisory Committees to assist with their decision-making process. The primary role of FDA Advisory Committee members is to provide the FDA with informed advice and recommendations on issues spanning science, regulatory policy, and the evaluation of products under the FDA’s jurisdiction. Although AdComm members serve the FDA in an advisory capacity, their recommendations are non-binding. Therefore, they do not have the final say in the regulatory approval process.
However, over the years, it has been made evident that the public is unaware of the role of Advisory Committees and ways in which they can engage with the FDA. In this case study, FAS hopes to share the current problem and actionable recommendations to combat public misconceptions regarding FDA AdComm roles and provide guidance on increasing FDA engagement with the public and other relevant stakeholders throughout the regulatory process.
Public Awareness Problems
While AdComm members are experts in their respective fields and volunteer their time to provide advice to the FDA, there are multiple factors that must be considered before making official decisions. The recommendations provided during Advisory Committee meetings are just one aspect that is considered for regulatory decision-making and do not guarantee an official approval or denial of a product by the FDA. During AdComm meetings, the FDA allows the general public to make public comments to the Agency and the AdComm regarding the topic that is being addressed. Despite this, members of the general public have expressed that, on many occasions, they are unaware AdComm meetings are occurring. This, in effect, deprives them of the opportunity to communicate directly with the FDA and the AdComm. Additionally, they feel the FDA fails to engage them in an adequate manner, thereby limiting opportunities for participatory engagement. It has also been noted that most members of the general public are unaware FDA Advisory Committees exist; and, for those who are aware, they are unclear about the capacity of their role within the regulatory process.
For these reasons, the FDA must take measures to enhance public understanding in an effort to combat misinformation, educate, and raise awareness on the existence of Committees and their purpose.
Communicating AdComms to the Public
Improving Public Awareness of Advisory Committees and their Role
Improving public awareness on the existence of FDA Advisory Committees and their purpose would assist the FDA with improving public trust and debunking myths and misinformation related to the approval of medical products. Advisory committees operate as an independent party and their recommendations assist with guiding regulatory decision-making. However, their recommendations are non-binding, and FDA leadership must consider additional factors before granting approvals or denials of medical products.
To increase public awareness on Advisory Committees, it should be made clear that AdComm recommendations are not conclusive, as the FDA considers multiple factors in its official decisions. The FDA can leverage social media platforms to increase awareness and understanding of AdComms through the use of disseminating information via the use of ads and active social media engagement. A survey conducted by Pew Research Center states that eight in ten Americans believe social media platforms are an effective way to bring awareness. In addition, disclaimers should be included on all public facing materials referencing AdComms to indicate their purpose. Clearly communicating this to the public will dispel myths that AdComms make the final call on the approvals of medical products.
Improving Communication about Advisory Committee Meetings
Encouraging public participation for Advisory Committee meetings will help foster a collaborative and engaged general public who can contribute valuable life experience to the regulatory process. FAS has identified ways in which the FDA can better communicate with the public to inform them of Advisory Committee meetings. First, the FDA can develop a webpage that allows people to receive notifications of upcoming AdComm meetings. The FDA can also establish relationships with state and local public health agencies, as well as advocacy organizations to spread awareness. Through these relationships, the various agencies and organizations can use their networks to disseminate widespread information on AdComm meetings. Public health agencies and advocacy organizations can gauge the best ways in which these communities would like the FDA to engage with them. This understanding of the communities they serve makes them an ideal partner for fostering continuous engagement.
Policy Recommendations
In an effort to improve public awareness and understanding of AdComms, the potential policy recommendations are as follows:
- FDA can leverage social media platforms to increase awareness and understanding of AdComms through the use of disseminating information (e.g., engagement, ads, etc.)
- FDA should include a disclaimer on all communications and marketing materials regarding AdComms
- These disclaimers should be made at all public meetings
- Disclaimers should emphasize the purpose of AdComm votes. Disclaimers should state that votes allow AdComm members to provide an official stance to the FDA as experts, but votes are non-binding.
- Develop a webpage that allows people to be placed on a listserv regarding upcoming meetings
- Partner with state and local public health agencies and advocacy organizations to spread awareness
Conclusion
Advisory Committees are essential to the FDA regulatory decision-making process. It’s imperative that their role is understood by them and the general public to best move the needle forward. While the FDA currently allows the public to provide public comment at Committee meetings, that alone cannot be considered engaging the community. The FDA must create new opportunities for interpersonal communication which will create an environment of mutual trust and understanding between both parties.